Biotech Accelerated: CRISPR and Beyond
1. Introduction to Genome Editing Technologies
1.1 Overview of Genome Editing: Concepts and Terminology
Genome editing refers to a set of techniques that allow scientists to make precise changes to the DNA sequence of an organism. Unlike traditional genetic modification, which often inserts foreign DNA randomly, genome editing targets specific locations in the genome to add, remove, or alter genetic material.
At its core, genome editing involves three main components:
- Target recognition: Identifying the exact DNA sequence to be edited.
- DNA cleavage: Creating a break in the DNA at the target site.
- DNA repair: Harnessing the cell’s natural repair mechanisms to introduce desired changes.
These steps are facilitated by engineered molecular tools, the most famous of which is CRISPR-Cas9.
Key Terms and Concepts
- Genome: The complete set of DNA in an organism, including all of its genes.
- Gene: A segment of DNA that codes for a protein or functional RNA.
- Nuclease: An enzyme that cuts DNA strands.
- Guide RNA (gRNA or sgRNA): A synthetic RNA molecule that directs nucleases like Cas9 to the target DNA sequence.
- Double-Strand Break (DSB): A break occurring simultaneously in both strands of the DNA helix.
- Non-Homologous End Joining (NHEJ): A DNA repair pathway that joins broken DNA ends directly, often causing small insertions or deletions.
- Homology-Directed Repair (HDR): A repair pathway that uses a homologous DNA template to accurately repair breaks, enabling precise edits.
Mind Map: Basic Genome Editing Workflow
Example: Editing a Gene to Knock Out Protein Function
Suppose researchers want to disable a gene responsible for producing a harmful protein. They design a guide RNA to direct Cas9 to the gene’s coding region. Cas9 creates a double-strand break, and the cell repairs it via NHEJ. This repair often introduces small insertions or deletions (indels) that disrupt the gene’s reading frame, effectively knocking out the protein.
Mind Map: Outcomes of DNA Repair After Editing
Example: Correcting a Disease-Causing Mutation
In diseases caused by a single faulty DNA base, HDR can be used to replace the mutation with the correct sequence. Researchers provide a DNA template alongside the editing machinery. After Cas9 cuts the DNA, the cell uses the template to repair the break, restoring the normal gene sequence.
Terminology Around Editing Tools
- CRISPR-Cas9: A bacterial immune system adapted for genome editing, where Cas9 is the nuclease guided by RNA.
- TALENs (Transcription Activator-Like Effector Nucleases): Engineered proteins that bind specific DNA sequences and induce breaks.
- ZFNs (Zinc Finger Nucleases): Custom DNA-binding proteins fused to nucleases for targeted cutting.
Each tool has strengths and weaknesses regarding specificity, ease of design, and delivery.
Mind Map: Genome Editing Tools
Summary
Genome editing is a precise method to alter DNA at chosen sites, relying on molecular tools to create breaks and cellular repair pathways to implement changes. Understanding the terminology and basic workflow is essential before exploring specific technologies and applications.
1.2 Historical Development of Genome Editing Tools
Genome editing is the process of making precise changes to the DNA sequence of an organism. The tools enabling this have evolved over decades, each generation improving on specificity, ease of use, and versatility. Understanding this history clarifies why CRISPR gained prominence and how earlier methods laid the groundwork.
Early Foundations: Restriction Enzymes and Recombinant DNA
In the 1970s, the discovery of restriction enzymes—molecular scissors that cut DNA at specific sequences—set the stage for genetic manipulation. These enzymes allowed scientists to cut and paste DNA fragments, leading to recombinant DNA technology. While powerful for cloning, this method lacked precision for targeted genome changes.
First Generation: Zinc Finger Nucleases (ZFNs)
ZFNs emerged in the 1990s as the first programmable nucleases. They combine a DNA-binding domain made of zinc finger proteins with a DNA-cleaving domain from the FokI endonuclease. Each zinc finger recognizes a 3-base pair DNA sequence, and by linking multiple fingers, researchers could target longer sequences.
Example: To disrupt a gene, two ZFNs bind opposite strands near the target site, and FokI domains dimerize to cut the DNA. The cell repairs the break, often introducing mutations that disable the gene.
Limitations: Designing zinc finger arrays is complex and costly. Off-target effects were a concern due to imperfect binding specificity.
Mind Map: Zinc Finger Nucleases
ZFN
├── DNA-binding domain
│ ├── Zinc finger motifs
│ └── Recognizes 3 bp per finger
├── Cleavage domain
│ └── FokI nuclease (requires dimerization)
├── Targeting
│ └── Custom assembly of zinc fingers
└── Applications
├── Gene disruption
└── Gene correction (limited)
Second Generation: Transcription Activator-Like Effector Nucleases (TALENs)
TALENs appeared in the late 2000s, inspired by bacterial proteins that bind DNA in a modular fashion. Each TAL effector repeat recognizes a single base pair, making design more straightforward than ZFNs.
Example: TALENs targeting the CCR5 gene in human cells demonstrated efficient gene disruption with fewer off-target effects compared to ZFNs.
Advantages: Easier to design and assemble; higher specificity.
Limitations: Large protein size complicates delivery into cells.
Mind Map: TALENs
TALEN
├── DNA-binding domain
│ ├── TALE repeats
│ └── Each repeat binds 1 bp
├── Cleavage domain
│ └── FokI nuclease (dimerizes)
├── Design
│ └── Modular assembly of repeats
└── Applications
├── Gene knockout
└── Targeted gene insertion
Third Generation: CRISPR-Cas Systems
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) systems were first characterized as a bacterial immune defense. The key innovation was using a guide RNA (gRNA) to direct the Cas nuclease to a complementary DNA sequence.
Example: CRISPR-Cas9 from Streptococcus pyogenes uses a single-guide RNA (sgRNA) to target DNA, inducing double-strand breaks that cells repair, enabling gene disruption or correction.
Advantages: Simple design (only the RNA sequence changes), high efficiency, multiplexing capability.
Limitations: Off-target effects and delivery challenges remain.
Mind Map: CRISPR-Cas9
CRISPR-Cas9
├── Components
│ ├── Cas9 nuclease
│ └── Single-guide RNA (sgRNA)
├── Targeting
│ └── RNA-DNA base pairing
├── Mechanism
│ └── Double-strand break induction
└── Applications
├── Gene knockout
├── Gene correction
└── Epigenetic modulation (with modified Cas9)
Other Genome Editing Tools
While ZFNs, TALENs, and CRISPR dominate, other tools like meganucleases and base editors have contributed to the field. Meganucleases are naturally occurring enzymes with long recognition sites, but their engineering is difficult. Base editors combine catalytically impaired Cas proteins with enzymes that chemically modify bases without cutting DNA, allowing precise nucleotide changes.
Summary Timeline
- 1970s: Restriction enzymes enable DNA cutting
- 1990s: Zinc Finger Nucleases introduce programmable targeting
- Late 2000s: TALENs improve ease of design and specificity
- 2012: CRISPR-Cas9 revolutionizes genome editing with RNA-guided targeting
Each step in this progression reflects a trade-off between complexity, specificity, and ease of use. The historical development shows a clear trend toward simpler, more flexible tools that empower a broader range of applications.
1.3 CRISPR-Cas Systems: Mechanisms and Variants
CRISPR-Cas systems are adaptive immune mechanisms originally discovered in bacteria and archaea. They protect these organisms from invading genetic elements like viruses and plasmids by recognizing and cutting foreign DNA. The system consists of two main components: the CRISPR array, which stores snippets of viral DNA as spacers, and the Cas (CRISPR-associated) proteins, which execute the targeting and cleavage of matching sequences.
Mechanism of CRISPR-Cas Systems
The CRISPR immune response unfolds in three stages:
- Adaptation: When a bacterium encounters foreign DNA, it integrates short fragments of this DNA into its CRISPR array as new spacers.
- Expression: The CRISPR array is transcribed into a long precursor RNA, which is processed into shorter CRISPR RNAs (crRNAs) containing individual spacer sequences.
- Interference: The crRNAs guide Cas proteins to complementary sequences in invading DNA, leading to targeted cleavage and neutralization.
This mechanism forms the basis for genome editing technologies, where the system is repurposed to target and modify specific DNA sequences in various organisms.
Key Variants of CRISPR-Cas Systems
CRISPR-Cas systems are diverse and classified into two classes and multiple types based on their protein components and mechanisms.
- Class 1: Multi-protein effector complexes (e.g., Type I and III systems).
- Class 2: Single, large Cas proteins that perform interference (e.g., Cas9, Cas12, Cas13).
The Class 2 systems are most commonly used in genome editing due to their simpler architecture.
Cas9
Cas9 is the most widely used nuclease. It requires two RNA components: a CRISPR RNA (crRNA) that matches the target DNA and a trans-activating crRNA (tracrRNA) that forms a complex with crRNA. In engineered systems, these are fused into a single guide RNA (sgRNA) for simplicity.
Cas9 recognizes a short sequence adjacent to the target called the protospacer adjacent motif (PAM), typically ‘NGG’ for Streptococcus pyogenes Cas9. Upon binding, Cas9 induces a double-strand break (DSB) three base pairs upstream of the PAM.
Example:
Editing the human EMX1 gene using SpCas9 involves designing an sgRNA complementary to the target site near an NGG PAM. Delivery of the Cas9-sgRNA complex into cells results in targeted DSBs that can be repaired to introduce mutations or insertions.
Cas12 (Cpf1)
Cas12 differs from Cas9 in several ways:
- It requires only a crRNA, no tracrRNA.
- Recognizes a T-rich PAM (e.g., ‘TTTV’).
- Produces staggered cuts with sticky ends rather than blunt ends.
These features can be advantageous for certain editing strategies.
Example:
Using Cas12a to target the CCR5 gene involves designing a crRNA matching the target sequence adjacent to a TTTV PAM. The staggered cut can facilitate precise insertions via homology-directed repair.
Cas13
Cas13 targets RNA instead of DNA, offering a tool for transient and reversible gene regulation or viral RNA targeting. It uses a crRNA to guide cleavage of complementary single-stranded RNA.
Example:
Cas13 can be programmed to degrade viral RNA in infected cells, reducing viral load without altering the host genome.
Mind Map: CRISPR-Cas System Components
Mind Map: Cas9 Mechanism
Mind Map: Differences Between Cas9 and Cas12
Practical Example: Designing a CRISPR Experiment with Cas9
Suppose you want to knock out the PCSK9 gene to study cholesterol metabolism. The steps include:
- Identify target sites in PCSK9 with NGG PAMs.
- Design sgRNAs complementary to these sites.
- Clone sgRNAs into expression vectors or synthesize as RNA.
- Deliver Cas9 and sgRNA into hepatocyte cell lines.
- Confirm editing by PCR and sequencing.
This example highlights the importance of PAM selection, guide design, and validation.
In summary, understanding the mechanisms and variants of CRISPR-Cas systems is essential for selecting the right tool for genome editing tasks. Cas9 remains the workhorse, but Cas12 and Cas13 offer alternative capabilities suited to specific applications.
1.4 Other Genome Editing Technologies: TALENs and ZFNs
While CRISPR-Cas systems have become the most widely used genome editing tools, two earlier technologies—TALENs (Transcription Activator-Like Effector Nucleases) and ZFNs (Zinc Finger Nucleases)—remain important for understanding the evolution of genome editing and for applications where CRISPR may not be ideal.
Zinc Finger Nucleases (ZFNs)
ZFNs are engineered proteins that combine a DNA-binding domain composed of zinc finger motifs with a DNA-cleaving domain, typically the FokI nuclease. Each zinc finger recognizes a 3-base pair DNA sequence, and multiple zinc fingers are linked to target longer sequences. When two ZFNs bind opposite strands of DNA at adjacent sites, the FokI domains dimerize and induce a double-strand break (DSB).
Key Features of ZFNs:
- Modular DNA recognition: Each zinc finger targets 3 base pairs, allowing flexible targeting by assembling multiple fingers.
- FokI nuclease domain: Requires dimerization, increasing specificity since two ZFNs must bind nearby.
- Custom design: Protein engineering is needed to create zinc finger arrays for new targets.
Example: Targeting the CCR5 Gene
ZFNs have been used to disrupt the CCR5 gene in human T cells to confer resistance to HIV infection. By designing zinc finger arrays that bind sequences flanking the CCR5 coding region, ZFNs induce DSBs that, when repaired by non-homologous end joining (NHEJ), disrupt gene function.
Mind Map: ZFN Structure and Function
Transcription Activator-Like Effector Nucleases (TALENs)
TALENs are similar in concept to ZFNs but use a different DNA-binding domain derived from transcription activator-like effectors (TALEs) found in Xanthomonas bacteria. Each TALE repeat recognizes a single base pair, and repeats are assembled to bind specific DNA sequences. Like ZFNs, TALENs use the FokI nuclease domain to create DSBs.
Key Features of TALENs:
- Single base recognition: Each TALE repeat targets one nucleotide, allowing straightforward design.
- FokI nuclease domain: Also requires dimerization for cleavage.
- Lower off-target effects: Compared to ZFNs, TALENs often show improved specificity.
Example: Correcting a Mutation in the Dystrophin Gene
TALENs have been applied to edit mutations in the dystrophin gene responsible for Duchenne muscular dystrophy. By designing TALEN pairs flanking the mutation site, targeted DSBs enable correction via homology-directed repair.
Mind Map: TALEN Design and Application
Comparison Between ZFNs and TALENs
| Feature | ZFNs | TALENs |
|---|---|---|
| DNA Recognition Unit | Zinc finger (3 bp per unit) | TALE repeat (1 bp per unit) |
| Design Complexity | Protein engineering required | Easier modular assembly |
| Targeting Range | Limited by finger context effects | More flexible targeting |
| Off-Target Effects | Higher risk | Generally lower |
| Size of Constructs | Smaller | Larger |
| Delivery | Easier due to smaller size | More challenging due to size |
Best Practices for Using TALENs and ZFNs
- Target Site Selection: Choose unique sequences to minimize off-target cleavage.
- Validation: Confirm cleavage efficiency and specificity using assays like T7E1 or deep sequencing.
- Delivery Method: Optimize delivery based on cell type; electroporation works well for TALENs despite their size.
- Control Experiments: Include non-targeting controls to assess background effects.
Practical Example: Using TALENs to Knock Out a Gene in Human Cells
- Identify target sequence within the gene.
- Design TALE repeats to bind sequences flanking the target site.
- Assemble TALEN constructs with FokI nuclease domains.
- Deliver TALEN plasmids into human cells via electroporation.
- Allow cells to repair DSBs, leading to indels and gene disruption.
- Validate knockout by PCR and sequencing.
This approach has been used successfully to disrupt genes like CCR5 and PD-1 in immune cells.
In summary, TALENs and ZFNs are protein-based genome editing tools that rely on engineered DNA-binding domains fused to nucleases. They require more complex protein design than CRISPR but offer alternative options when CRISPR is less suitable. Both have demonstrated utility in research and therapeutic contexts, with clear protocols and examples to guide their application.
1.5 Best Practices in Selecting Genome Editing Tools with Practical Examples
Selecting the right genome editing tool is a foundational step in any project involving genetic modification. The choice influences efficiency, specificity, ease of use, and ultimately the success of your experiment or therapeutic design. This section outlines best practices for selecting genome editing tools, supported by practical examples and mind maps to clarify decision-making.
Understanding Your Project Requirements
Before choosing a tool, clearly define your objectives. Are you aiming for a simple gene knockout, precise base editing, or transcriptional regulation? The target organism and cell type also matter, as delivery methods and editing efficiencies vary.
Key Factors to Consider
- Specificity: How precise must the edit be? Some tools have higher off-target risks.
- Efficiency: What level of editing efficiency is acceptable?
- Delivery: Which delivery methods are compatible with your cells?
- Target Sequence Constraints: Does the target site have PAM or other sequence requirements?
- Type of Edit: Knockout, knock-in, base editing, or epigenetic modulation?
- Tool Complexity and Resources: Availability of reagents, expertise, and cost.
Mind Map: Tool Selection Criteria
Comparing Common Genome Editing Tools
| Tool | Specificity | Efficiency | Delivery Options | Target Constraints | Typical Use Cases |
|---|---|---|---|---|---|
| CRISPR-Cas9 | Moderate | High | Viral, Electroporation, Lipid Nanoparticles | NGG PAM site required | Gene knockout, knock-in |
| Cas12a | Higher | Moderate | Similar to Cas9 | TTTV PAM site required | Multiplex editing, staggered cuts |
| TALENs | High | Moderate | Electroporation, Viral | No strict PAM, but target design complex | Precise editing, hard-to-target sites |
| ZFNs | High | Variable | Electroporation, Viral | Target site dependent | Early gene editing, therapeutic applications |
Practical Example 1: Choosing Between CRISPR-Cas9 and Cas12a for Plant Editing
A research team wants to edit a gene in rice to improve drought resistance. The target site has a TTTV PAM but no NGG PAM nearby. Cas12a is a better fit due to its PAM requirement, despite slightly lower efficiency. Delivery is via Agrobacterium-mediated transformation, compatible with both tools.
Practical Example 2: Selecting TALENs for a Difficult Target in Human Cells
A lab aims to disrupt a gene with a highly repetitive sequence where CRISPR guide design is challenging. TALENs, which rely on protein-DNA recognition rather than RNA guides, provide higher specificity and fewer off-target effects in this context, despite more complex assembly.
Mind Map: Decision Flow for Tool Selection
Practical Example 3: Delivery Constraints Dictate Tool Choice
In primary neurons, viral delivery is limited due to toxicity. Electroporation is possible but harsh. Lipid nanoparticles are preferred. CRISPR-Cas9 RNP complexes delivered via lipid nanoparticles offer transient editing with reduced off-target effects, making them suitable here.
Summary of Best Practices
- Define your editing goal clearly: Different tools excel at different types of edits.
- Analyze target sequence constraints: PAM availability can rule in or out certain nucleases.
- Match delivery methods to cell type: Some tools are easier to deliver in certain contexts.
- Balance specificity and efficiency: Higher specificity often means more complex design or lower efficiency.
- Consider resource availability: Choose tools that fit your lab’s expertise and budget.
- Use decision flowcharts or mind maps: Visualizing choices helps avoid oversight.
Selecting the right genome editing tool is a matter of matching your project’s unique needs with the strengths and limitations of available technologies. Practical examples show that no single tool fits all scenarios; thoughtful evaluation leads to better outcomes.
2. CRISPR-Cas Systems in Detail
2.1 Structure and Function of CRISPR-Cas9
CRISPR-Cas9 is a genome editing tool derived from a bacterial adaptive immune system. Its core function is to locate and cut specific DNA sequences, enabling targeted genetic modifications. Understanding its structure and function is key to applying it effectively.
Components of CRISPR-Cas9
- Cas9 Protein: An endonuclease enzyme that cuts DNA at specific sites.
- Guide RNA (gRNA): A synthetic RNA molecule that directs Cas9 to the target DNA sequence.
- Protospacer Adjacent Motif (PAM): A short DNA sequence next to the target site, essential for Cas9 recognition.
How CRISPR-Cas9 Works
- The guide RNA binds to the complementary DNA sequence.
- Cas9 scans the DNA for a PAM sequence (usually 5’-NGG-3’ for the common SpCas9).
- Upon PAM recognition, Cas9 unwinds the DNA and checks for complementarity with the guide RNA.
- If matched, Cas9 induces a double-strand break (DSB) three base pairs upstream of the PAM.
Mind Map: CRISPR-Cas9 Structure and Function
Cas9 Protein Domains
Cas9 has two nuclease domains: HNH and RuvC. The HNH domain cleaves the DNA strand complementary to the guide RNA, while RuvC cleaves the opposite strand. Together, they create a double-strand break. The PAM recognition domain ensures Cas9 only cuts DNA next to the PAM sequence, preventing unwanted cuts.
Guide RNA Design
The guide RNA is typically engineered as a single guide RNA (sgRNA) combining crRNA and tracrRNA. The 20-nucleotide sequence at the 5’ end of the sgRNA matches the target DNA. This sequence must be chosen carefully to avoid off-target effects.
Example: Editing the Human EMX1 Gene
Suppose you want to edit the EMX1 gene in human cells. You design an sgRNA with a 20-nt sequence complementary to a unique region in EMX1. The target site must be adjacent to a PAM (NGG). Cas9 binds the sgRNA, scans the genome, finds the PAM, and checks for complementarity. Once confirmed, it cuts the DNA, triggering repair mechanisms that can introduce mutations or insertions.
Mind Map: CRISPR-Cas9 Targeting Process
Cellular Repair After Cas9 Cleavage
After Cas9 cuts the DNA, the cell repairs the break. The two main pathways are:
- Non-Homologous End Joining (NHEJ): Joins DNA ends directly, often causing insertions or deletions (indels) that can disrupt gene function.
- Homology-Directed Repair (HDR): Uses a homologous DNA template to repair accurately, enabling precise edits if a donor template is provided.
Example: Creating a Gene Knockout
Using NHEJ, a double-strand break in a coding exon can cause frameshift mutations, effectively knocking out the gene. For instance, targeting the CCR5 gene in immune cells with CRISPR-Cas9 can disrupt its function, which has been explored in HIV research.
Summary
CRISPR-Cas9 is a programmable nuclease system where the Cas9 protein, guided by an RNA molecule, locates and cleaves DNA at specific sites defined by the guide sequence and PAM. Its modular structure and straightforward targeting mechanism make it a powerful tool for genome editing.
2.2 Alternative CRISPR Systems: Cas12, Cas13 and Beyond
CRISPR systems have expanded beyond the well-known Cas9, introducing new enzymes like Cas12 and Cas13 that offer distinct mechanisms and applications. Understanding these alternatives helps tailor genome editing approaches to specific needs.
Cas12: DNA Targeting with a Twist
Cas12, originally termed Cpf1, is a single RNA-guided endonuclease that targets double-stranded DNA but differs from Cas9 in several key ways:
- Cas12 requires a T-rich PAM (Protospacer Adjacent Motif), typically TTTV (where V is A, C, or G), unlike Cas9’s G-rich PAM.
- It creates staggered cuts with 5’ overhangs instead of blunt ends, which can facilitate certain DNA insertions.
- Cas12 processes its own CRISPR RNA array, simplifying multiplex targeting.
Mind Map: Cas12 Features
Example: Using Cas12 for Multiplex Editing
A lab aiming to knock out several genes in a plant species used Cas12 due to its ability to process multiple crRNAs from a single transcript. This reduced the complexity of vector design and improved editing efficiency compared to Cas9, which requires separate sgRNAs.
Cas13: RNA Targeting Enzyme
Cas13 is unique in that it targets single-stranded RNA rather than DNA. This opens possibilities for transient gene regulation and RNA virus targeting.
- Cas13 uses a guide RNA to bind complementary RNA sequences.
- Upon target recognition, Cas13 exhibits collateral cleavage activity, cutting nearby RNAs nonspecifically.
- Different Cas13 variants (Cas13a, Cas13b, Cas13d) vary in size and activity.
Mind Map: Cas13 Characteristics
Example: RNA Knockdown in Mammalian Cells
Researchers used Cas13d to selectively degrade transcripts of a disease-causing gene in human cells. Because Cas13 targets RNA, the effect was reversible and did not alter the genome, offering a safer alternative for transient gene silencing.
Beyond Cas12 and Cas13: Other CRISPR Systems
Several other CRISPR effectors have been characterized, each with unique properties:
- Cas14: A small nuclease targeting single-stranded DNA, useful for diagnostics.
- Cas3: Part of Type I systems, capable of long-range DNA degradation rather than precise cuts.
Mind Map: Other CRISPR Effectors
Practical Considerations
Choosing between Cas9, Cas12, Cas13, or other systems depends on the target molecule (DNA vs RNA), desired cut pattern, and application (permanent edit vs transient modulation).
- For precise DNA edits with sticky ends, Cas12 is advantageous.
- For RNA targeting without genome modification, Cas13 is preferred.
- For multiplex editing with simpler guide arrays, Cas12’s autonomous processing is useful.
Summary Table
| System | Target | PAM Requirement | Cleavage Type | Unique Feature |
|---|---|---|---|---|
| Cas9 | dsDNA | G-rich (NGG) | Blunt ends | Widely used, well-characterized |
| Cas12 | dsDNA | T-rich (TTTV) | 5’ overhangs | Processes own crRNA, sticky ends |
| Cas13 | ssRNA | None | Collateral cleavage | RNA targeting, transient effects |
| Cas14 | ssDNA | None | Collateral cleavage | Very small size |
| Cas3 | dsDNA | Varies | Processive degradation | Large deletions |
This overview highlights how alternative CRISPR systems expand the toolkit for genome and transcriptome engineering, each with distinct properties suited to different experimental goals.
2.3 Guide RNA Design: Principles and Tools
Guide RNA (gRNA) is the navigator for CRISPR-Cas systems, directing the nuclease to the precise DNA sequence to be edited. Designing an effective gRNA is crucial for achieving high editing efficiency and minimizing off-target effects. This section covers the core principles of gRNA design, common pitfalls, and practical examples to illustrate best practices.
Core Principles of Guide RNA Design
- Target Specificity: The gRNA sequence must uniquely match the target DNA region to avoid unintended cuts elsewhere in the genome.
- PAM Recognition: The protospacer adjacent motif (PAM) is a short DNA sequence immediately following the target site, required for Cas binding. Different Cas proteins recognize different PAMs (e.g., NGG for SpCas9).
- GC Content: Optimal GC content (typically 40-60%) balances binding stability and flexibility. Too low GC content weakens binding; too high can cause secondary structures.
- Avoidance of Secondary Structures: gRNAs that form hairpins or other structures can reduce Cas9 binding and cleavage efficiency.
- Position Relative to Functional Domains: For gene knockout, targeting early exons can increase the chance of disrupting protein function.
Mind Map: Guide RNA Design Considerations
Step-by-Step Guide RNA Design Process
- Identify the Target Region: Choose the gene or locus to edit. For knockouts, early exons are preferred.
- Locate PAM Sites: Scan the target region for PAM sequences compatible with the chosen Cas protein.
- Select Candidate gRNAs: Extract sequences adjacent to PAM sites (usually 20 nucleotides for SpCas9).
- Evaluate GC Content: Calculate GC percentage; discard candidates outside the optimal range.
- Check for Secondary Structures: Use RNA folding tools to predict and exclude gRNAs prone to hairpins.
- Assess Off-Target Potential: Compare candidate sequences against the genome to identify potential off-target matches.
- Prioritize Candidates: Based on specificity scores, predicted efficiency, and experimental context.
Mind Map: Guide RNA Design Workflow
Examples of Guide RNA Design
Example 1: Designing a gRNA for Human Beta-Globin Gene (HBB) Knockout
- Target: Exon 1 of HBB gene.
- PAM: NGG (SpCas9).
- Candidate sequence near PAM: 5’-GAGTCTGAGTCTGCCGTTACTGG-3’ (PAM underlined: TGG).
- Extract 20 nt guide: GAGTCTGAGTCTGCCGTTACT.
- GC content: 11 G/C out of 20 = 55% (within optimal range).
- RNA folding prediction: Minimal secondary structure.
- Off-target check: No perfect matches elsewhere; 1 mismatch in non-coding region.
This gRNA is a strong candidate for efficient and specific editing.
Example 2: Avoiding a gRNA with High Off-Target Risk
- Candidate sequence: 5’-GGGCGGCGGCGGCGGCGGCG-3’.
- GC content: 95% (too high).
- Secondary structure prediction: Strong hairpin formation.
- Off-target analysis: Multiple perfect matches in repetitive genomic regions.
This gRNA should be discarded due to poor specificity and structural issues.
Tools and Techniques for gRNA Design
While this section focuses on principles, practical design often involves computational tools that automate many steps. These tools typically incorporate genome-wide off-target prediction, efficiency scoring, and secondary structure analysis. When using such tools, understanding the underlying principles helps interpret their output critically.
Summary
Effective gRNA design balances specificity, efficiency, and practical considerations like PAM compatibility and target location. By following a structured workflow and evaluating candidates carefully, researchers can improve editing outcomes and reduce unintended effects. Concrete examples highlight how these principles apply in real scenarios, making the design process clearer and more manageable.
2.4 Delivery Methods for CRISPR Components: Viral and Non-Viral
Delivery Methods for CRISPR Components: Viral and Non-Viral
Delivering CRISPR components efficiently into target cells is a critical step in genome editing. The choice of delivery method affects editing efficiency, cell viability, and off-target effects. Broadly, delivery approaches fall into two categories: viral and non-viral. Each has distinct advantages and limitations, which we will explore alongside practical examples and mind maps to clarify their relationships.
Viral Delivery Methods
Viral vectors exploit the natural ability of viruses to enter cells and deliver genetic material. Common viral vectors for CRISPR delivery include lentivirus, adenovirus, and adeno-associated virus (AAV).
-
Lentivirus: Integrates into the host genome, enabling stable, long-term expression of CRISPR components. Useful for editing dividing and non-dividing cells but carries risks of insertional mutagenesis.
-
Adenovirus: Does not integrate into the genome, leading to transient expression. It has a large packaging capacity but can trigger strong immune responses.
-
Adeno-Associated Virus (AAV): Offers low immunogenicity and mostly remains episomal, providing transient expression. Its limited packaging size (~4.7 kb) can restrict delivery of large CRISPR constructs.
Mind Map: Viral Delivery Methods
Example: To edit human T cells for CAR-T therapy, lentiviral vectors are often used because they provide stable integration of the CAR transgene, ensuring persistent expression. However, for in vivo editing of retinal cells, AAV vectors are preferred due to their low immunogenicity and ability to infect non-dividing cells.
Non-Viral Delivery Methods
Non-viral methods avoid the risks associated with viral vectors but often face challenges in delivery efficiency and cell toxicity. They include physical methods, chemical carriers, and direct delivery of ribonucleoprotein complexes (RNPs).
-
Electroporation: Uses electrical pulses to transiently permeabilize cell membranes, allowing CRISPR components (DNA, RNA, or RNPs) to enter. Highly efficient in many cell types but can cause cell death if parameters are not optimized.
-
Lipid Nanoparticles (LNPs): Lipid-based vesicles encapsulate CRISPR components, facilitating cellular uptake via endocytosis. LNPs are less toxic than electroporation and suitable for in vivo delivery.
-
Microinjection: Direct injection of CRISPR components into cells or embryos. Precise but low throughput and technically demanding.
-
Ribonucleoprotein (RNP) Delivery: Direct delivery of Cas9 protein complexed with guide RNA. Offers transient activity, reducing off-target effects and immune responses.
Mind Map: Non-Viral Delivery Methods
Example: Editing primary human hematopoietic stem cells (HSCs) often uses electroporation to deliver Cas9 RNPs. This approach achieves high editing efficiency while minimizing prolonged Cas9 expression, which reduces off-target risks.
Comparing Viral and Non-Viral Delivery
| Feature | Viral Delivery | Non-Viral Delivery |
|---|---|---|
| Expression Duration | Stable (lentivirus) or transient (AAV, adenovirus) | Transient (RNPs, LNPs, electroporation) |
| Packaging Capacity | Large (adenovirus, lentivirus), limited (AAV) | Limited by carrier type |
| Immunogenicity | Can be high (adenovirus) | Generally lower |
| Integration Risk | Yes (lentivirus) | No |
| Delivery Efficiency | High | Variable, often lower |
| Cell Type Compatibility | Broad | Depends on method |
Practical Considerations
-
Cell Type: Hard-to-transfect cells like primary neurons may require viral vectors or optimized electroporation.
-
Editing Goal: For transient editing and reduced off-target effects, RNP delivery via electroporation or LNPs is preferred.
-
Safety: Clinical applications often avoid integrating vectors to reduce mutagenesis risk.
-
Scale: Viral vectors are scalable for clinical manufacturing but require complex production.
Mind Map: Delivery Method Selection Factors
Summary Example: Editing Sickle Cell Disease HSCs
In a clinical trial for sickle cell disease, CRISPR-Cas9 RNPs are delivered into patient-derived HSCs via electroporation. This non-viral method ensures transient Cas9 activity, reducing off-target effects and avoiding genomic integration. After editing, cells are expanded and infused back into the patient. This example illustrates how delivery method choice aligns with safety, efficiency, and therapeutic goals.
In conclusion, selecting a delivery method for CRISPR components requires balancing efficiency, safety, and the biological context. Viral vectors offer high efficiency and stable expression but carry integration and immunogenicity risks. Non-viral methods provide transient editing with lower risks but may require optimization for each cell type and application.
2.5 Case Study: Efficient CRISPR-Cas9 Editing in Human Cell Lines
This case study walks through a practical example of using CRISPR-Cas9 to edit a gene in a human cell line, focusing on the workflow, challenges, and best practices. The goal is to introduce a targeted knockout of the TP53 gene in HEK293T cells, a widely used human embryonic kidney cell line.
Step 1: Define the Objective and Target
The TP53 gene encodes the p53 protein, a tumor suppressor involved in DNA repair and apoptosis. Knocking out TP53 allows researchers to study cell cycle regulation and cancer biology. The first step is to select a target region within TP53 that, when disrupted, will likely cause loss of function.
- Target exon: Exon 4, chosen because it encodes part of the DNA-binding domain.
- Desired edit: Frameshift mutation causing premature stop codon.
Step 2: Guide RNA (gRNA) Design
Designing an effective gRNA is critical. The criteria include:
- High on-target activity score.
- Minimal predicted off-target sites.
- Targeting early exons to maximize knockout effect.
Using a gRNA design tool, two candidate gRNAs were selected:
- gRNA1: 5’-GGAGGTTGTGAGGCGCTGTC-3’
- gRNA2: 5’-CAGGCTCCCAGAGACCTGTA-3’
Both target exon 4 but differ in predicted off-target profiles.
Step 3: Cloning and Validation of gRNA Constructs
The gRNA sequences were cloned into a plasmid expressing Cas9 and a puromycin resistance marker. Cloning was verified by Sanger sequencing.
Step 4: Delivery to HEK293T Cells
HEK293T cells were cultured under standard conditions. Transfection was performed using lipofection, a common non-viral method suitable for this cell line.
- Transfection reagent: Lipofectamine 3000.
- DNA amount: 2 µg per well (6-well plate).
- Controls: Non-targeting gRNA and mock transfection.
Step 5: Selection and Expansion
After 48 hours, puromycin was added to select for successfully transfected cells. Selection continued for 3 days, after which surviving cells were expanded.
Step 6: Screening for Editing Efficiency
Bulk population genomic DNA was extracted. Editing efficiency was assessed by:
- T7 Endonuclease I assay: Detects mismatches from indels.
- Sanger sequencing followed by ICE (Inference of CRISPR Edits) analysis.
Results showed ~65% editing efficiency with gRNA1 and ~50% with gRNA2.
Step 7: Isolation of Clonal Cell Lines
To obtain pure knockout lines, single cells were sorted by flow cytometry into 96-well plates. Clones were expanded and genotyped.
- PCR amplification of target locus.
- Sanger sequencing to confirm frameshift mutations.
Out of 24 clones, 10 showed biallelic frameshift mutations with gRNA1.
Step 8: Functional Validation
Loss of p53 protein was confirmed by Western blot. Functional assays included:
- Cell cycle analysis by flow cytometry, showing altered G1/S checkpoint.
- Increased resistance to DNA-damaging agents, consistent with TP53 loss.
Mind Map: CRISPR-Cas9 Editing Workflow in Human Cell Lines
Best Practices Highlighted
- Target early exons to maximize knockout likelihood.
- Use multiple gRNAs to compare efficiency and specificity.
- Include proper controls such as non-targeting gRNAs and mock transfections.
- Validate constructs by sequencing before transfection.
- Select for transfected cells to enrich edited populations.
- Screen bulk populations first to estimate editing efficiency.
- Isolate clonal lines for downstream functional studies.
- Confirm edits at DNA and protein levels.
Example: Off-Target Analysis
To assess off-target effects, the top 5 predicted off-target sites for gRNA1 were PCR amplified and sequenced. No indels were detected, indicating high specificity in this context.
Summary
This case study demonstrates a straightforward approach to knocking out a gene in human cells using CRISPR-Cas9. It emphasizes careful design, validation, and functional testing. The workflow and practices shown here can be adapted to other genes and cell types with appropriate modifications.
3. Genome Editing Workflow and Experimental Design
3.1 Defining Objectives and Target Selection
Before starting any genome editing experiment, the first step is to clearly define what you want to achieve. This sounds obvious, but it’s easy to overlook the importance of a precise objective. The objective guides every subsequent decision, from choosing the target gene to selecting the editing strategy and delivery method.
Why Define Objectives Clearly?
- Sets measurable goals.
- Helps prioritize targets.
- Guides experimental design and controls.
- Facilitates interpretation of results.
Common Types of Objectives
- Gene knockout: Disable a gene to study its function.
- Gene correction: Fix a mutation causing disease.
- Gene insertion: Add a new gene or regulatory element.
- Gene regulation: Modify expression levels without changing the sequence.
Mind Map: Defining Objectives
Once the objective is clear, the next step is target selection. This involves choosing the specific DNA sequence or gene region to edit.
Target Selection
Target selection depends on the objective and the biological context. It requires understanding the gene’s structure, function, and relevance to the system or disease studied.
Key considerations include:
- Gene function: Is the gene essential? What phenotype results from its alteration?
- Target region: Coding sequence, promoter, enhancer, splice sites?
- Sequence accessibility: Chromatin state can affect editing efficiency.
- Off-target risk: Similar sequences elsewhere in the genome.
- Delivery feasibility: Some targets may be easier to reach depending on cell type.
Mind Map: Target Selection Factors
Example 1: Knockout of a Metabolic Enzyme in Liver Cells
Objective: Understand the role of enzyme X in liver metabolism.
Target Selection:
- Choose an early exon to ensure complete loss of function.
- Verify the exon is unique to avoid off-target effects.
- Confirm the region is accessible in hepatocytes.
Outcome: Designing sgRNAs targeting exon 2 of gene X, with minimal predicted off-target sites.
Example 2: Correcting a Point Mutation in a Blood Disorder
Objective: Repair a single nucleotide mutation causing sickle cell disease.
Target Selection:
- Target the exact mutation site in the beta-globin gene.
- Consider homology-directed repair (HDR) for precise correction.
- Evaluate nearby PAM sites for CRISPR-Cas9 compatibility.
Outcome: Selecting sgRNAs flanking the mutation and designing a repair template.
Practical Tips
- Start with a literature review to understand gene function and known variants.
- Use genome browsers to visualize gene structure and regulatory elements.
- Employ computational tools to predict off-target effects early.
- Consider multiple target sites to increase chances of success.
- Align your target choice with your delivery method and cell type.
Summary
Defining your objective and selecting the right target are foundational steps in genome editing. Clear objectives prevent wasted effort, and thoughtful target selection maximizes editing efficiency and relevance. Mind maps can help organize these considerations visually, ensuring no critical factor is missed.
3.2 Designing sgRNAs: Specificity and Off-Target Considerations
Single guide RNAs (sgRNAs) are the navigators of the CRISPR-Cas9 system, directing the Cas9 nuclease to precise DNA sequences. Designing sgRNAs with high specificity is crucial to ensure efficient editing and to minimize unintended changes elsewhere in the genome. This section covers the principles behind sgRNA design, factors influencing specificity, and practical examples illustrating these concepts.
Understanding sgRNA Structure and Targeting
An sgRNA typically consists of a 20-nucleotide sequence complementary to the target DNA, followed by a scaffold region that binds Cas9. The target DNA must be adjacent to a protospacer adjacent motif (PAM), usually ‘NGG’ for SpCas9.
Key points:
- The 20-nt guide sequence determines target specificity.
- PAM presence is mandatory for Cas9 binding and cleavage.
- Mismatches between sgRNA and DNA can reduce binding and cutting efficiency but may also cause off-target effects if tolerated.
Mind Map: sgRNA Design Factors
Specificity and Off-Target Considerations
Specificity depends on how well the sgRNA matches only the intended target. Off-target effects arise when the sgRNA binds and directs Cas9 to similar but unintended sequences, potentially causing unwanted mutations.
- Seed Region: The first 8-12 nucleotides next to the PAM are critical. Mismatches here drastically reduce binding.
- Mismatch Position: Mismatches near the PAM are less tolerated than those farther away.
- Number of Mismatches: More mismatches generally reduce off-target binding, but some positions are more forgiving.
Mind Map: Off-Target Factors
Practical Example 1: Designing an sgRNA for the Human HBB Gene
Suppose you want to edit the HBB gene, responsible for beta-globin production. The target site is within exon 1.
- Identify PAM sites near the mutation.
- Select 20-nt sequences upstream of PAM.
- Check GC content; aim for 40-60% to balance stability and binding.
- Use in silico tools to scan the genome for similar sequences with fewer than 3 mismatches.
- Prioritize guides with minimal predicted off-targets, especially in coding regions.
For example, an sgRNA targeting 5’-GAGTCTGCCGTTACTGCCCT-3’ adjacent to ‘NGG’ PAM has 50% GC content and no perfect off-target matches.
Practical Example 2: Minimizing Off-Target Effects by Truncated sgRNAs
Studies show that shortening the guide sequence from 20 to 17-18 nucleotides can reduce off-target cleavage without severely compromising on-target efficiency.
- Design a truncated sgRNA targeting the same site.
- Test both full-length and truncated guides in vitro.
- Compare editing efficiency and off-target activity using sequencing.
This approach can be particularly useful when off-target sites have only one or two mismatches.
Additional Design Tips
- Avoid sgRNAs with homopolymer runs (e.g., four or more identical nucleotides in a row).
- Consider chromatin state; open chromatin regions are more accessible.
- Use chemical modifications on sgRNAs to improve stability and reduce immune responses.
Mind Map: sgRNA Optimization Workflow
In summary, designing sgRNAs requires balancing efficiency and specificity. Careful selection of target sites, awareness of mismatch tolerance, and validation through experiments help ensure successful genome editing with minimal unintended effects.
3.3 Construct Assembly and Validation
Construct assembly is the process of creating the DNA plasmid or vector that carries the genome editing components, such as the Cas nuclease and guide RNA (gRNA) sequences. Validation ensures the construct is correct and functional before moving to cell delivery. This section breaks down the steps and considerations for assembling and validating genome editing constructs.
Key Steps in Construct Assembly
- Designing the Insert: Choose the gRNA sequence targeting your gene of interest. Incorporate necessary promoters, terminators, and selection markers.
- Vector Selection: Pick a backbone compatible with your delivery method and host cells. Consider size, copy number, and antibiotic resistance.
- Cloning Strategy: Decide between restriction enzyme cloning, Gibson assembly, Golden Gate assembly, or other methods based on complexity and resources.
- Transformation and Screening: Introduce the assembled plasmid into bacteria, then screen colonies for correct inserts.
Mind Map: Construct Assembly Workflow
Cloning Strategies Explained
- Restriction Enzyme Cloning: Traditional method using restriction sites flanking your insert and vector. Reliable but can be limited by available sites.
- Gibson Assembly: Seamless joining of multiple DNA fragments with overlapping ends in a single reaction. Useful for complex constructs.
- Golden Gate Assembly: Uses type IIS restriction enzymes to assemble multiple fragments directionally and efficiently.
Example: Cloning a gRNA into a CRISPR-Cas9 Vector Using Golden Gate Assembly
- Design oligonucleotides encoding the gRNA target sequence with compatible overhangs.
- Anneal oligos to form double-stranded DNA.
- Mix annealed oligos with the vector backbone, type IIS enzyme (e.g., BsaI), and ligase.
- Incubate in a thermocycler cycling between digestion and ligation temperatures.
- Transform competent E. coli and plate on selective media.
- Screen colonies by colony PCR or restriction digest.
- Confirm sequence by Sanger sequencing.
Validation Techniques
- Colony PCR: Quick check for insert presence and size.
- Restriction Digest Analysis: Confirms insert orientation and integrity.
- Sanger Sequencing: Gold standard for verifying exact sequence, especially critical for gRNA regions.
- Functional Assays: Testing expression of Cas protein and gRNA in cells or in vitro cleavage assays.
Mind Map: Construct Validation Methods
Practical Example: Validating a CRISPR Construct Targeting the HBB Gene
After cloning the gRNA targeting the HBB gene into a Cas9 vector:
- Perform colony PCR using primers flanking the gRNA insertion site. Expect a band size corresponding to the vector plus gRNA insert.
- Digest plasmid DNA from positive colonies with enzymes cutting within the insert and vector to confirm orientation.
- Sequence the gRNA region to verify no mutations were introduced during cloning.
- Transfect the validated plasmid into HEK293 cells and perform a T7 endonuclease assay to detect cleavage at the HBB target site.
This stepwise validation ensures the construct is both correct at the sequence level and functional in cells.
Tips and Best Practices
- Use high-fidelity polymerases during PCR to minimize mutations.
- Always sequence the entire gRNA region, including promoter and scaffold sequences.
- Include negative controls in cloning and validation steps to identify background signals.
- Maintain sterile technique to avoid contamination during bacterial transformations.
- Document each step carefully to track construct versions and modifications.
Construct assembly and validation are foundational to successful genome editing experiments. Taking time to confirm the accuracy and functionality of your constructs reduces downstream troubleshooting and increases confidence in your results.
3.4 Delivery Optimization for Different Cell Types
Delivering genome editing tools efficiently and safely into cells is a key step in any editing experiment. Different cell types respond differently to delivery methods, so optimization is essential. This section covers common delivery approaches, challenges specific to various cell types, and practical examples to guide your choices.
Delivery Methods Overview
- Viral vectors: Efficient for many cell types, especially hard-to-transfect cells, but can raise safety and immunogenicity concerns.
- Electroporation: Uses electric pulses to create temporary pores in the membrane; effective but can cause cell stress.
- Lipid nanoparticles (LNPs): Non-viral, less toxic, suitable for many cell types but sometimes less efficient.
- Physical methods: Microinjection and biolistics, typically for specialized applications.
Mind Map: Delivery Method Selection Factors
Cell Type Considerations
-
Primary Cells
- Often difficult to transfect.
- Electroporation of RNP complexes is common.
- Viral vectors (lentivirus, AAV) can be effective but require biosafety measures.
- Example: Human T cells respond well to electroporation using optimized pulse conditions.
-
Stem Cells (e.g., iPSCs, ESCs)
- Sensitive to stress; viability is a concern.
- Nucleofection (a form of electroporation) is widely used.
- RNP delivery reduces prolonged nuclease expression, lowering off-target risks.
- Example: iPSCs edited via nucleofection with Cas9 RNP show high editing efficiency and maintain pluripotency.
-
Immortalized Cell Lines (e.g., HEK293, HeLa)
- Generally easier to transfect.
- Lipid-based transfection reagents work well.
- Plasmid DNA delivery is common.
- Example: HEK293 cells transfected with plasmid encoding Cas9 and sgRNA using lipofection achieve >80% editing.
-
Suspension vs Adherent Cells
- Suspension cells often require electroporation or viral delivery.
- Adherent cells can be transfected with lipids or electroporation.
Mind Map: Delivery Optimization Workflow
Practical Example: Optimizing Electroporation in Primary Human T Cells
- Objective: Deliver Cas9 RNP to knock out a target gene.
- Initial Approach: Use standard electroporation settings from literature.
- Observed Issues: High cell death, moderate editing.
- Optimization Steps:
- Adjust pulse voltage and duration to reduce toxicity.
- Test different RNP concentrations to balance efficiency and viability.
- Include recovery media with cytokines post-electroporation.
- Outcome: Achieved 70% editing with >80% viability.
Practical Example: Lipid Nanoparticle Delivery in Hepatocytes
- Objective: Deliver mRNA encoding Cas9 and sgRNA.
- Challenges: Primary hepatocytes are sensitive and difficult to transfect.
- Optimization Steps:
- Screen different lipid formulations.
- Optimize lipid-to-mRNA ratio.
- Minimize incubation time to reduce toxicity.
- Outcome: Moderate editing (~50%) with acceptable viability.
Tips for Successful Delivery Optimization
- Always include controls: untreated cells, mock transfection.
- Monitor both editing efficiency and cell viability.
- Consider delivery format: RNPs often yield faster editing and lower off-target effects.
- Use small-scale pilot experiments before scaling up.
- Document all parameters for reproducibility.
Summary
Optimizing delivery requires understanding your cell type’s characteristics and balancing efficiency with viability. Iterative testing and parameter tuning are necessary. Using appropriate delivery formats and methods tailored to the cell type will improve your editing outcomes.
3.5 Practical Example: Editing a Disease-Associated Gene in Stem Cells
This section walks through a practical example of editing a gene linked to a genetic disorder in human induced pluripotent stem cells (iPSCs). The goal is to correct a point mutation in the HBB gene, which causes beta-thalassemia, a blood disorder characterized by reduced hemoglobin production.
Step 1: Define the Objective and Target
The HBB gene mutation targeted is a single nucleotide substitution known to disrupt normal hemoglobin function. The objective is to use CRISPR-Cas9 to replace the mutated base with the wild-type sequence in patient-derived iPSCs.
Mind Map: Objective and Target Definition
Step 2: Design of sgRNA and Donor Template
A single guide RNA (sgRNA) is designed to bind near the mutation site, maximizing on-target activity and minimizing off-target effects. Alongside, a single-stranded oligodeoxynucleotide (ssODN) donor template is synthesized to provide the correct nucleotide sequence for homology-directed repair (HDR).
Key considerations:
- sgRNA target sequence must be adjacent to a PAM site (NGG for SpCas9).
- Avoid sgRNAs with predicted off-targets in coding regions.
- Donor template includes silent mutations to prevent re-cutting post-editing.
Mind Map: sgRNA and Donor Design
Step 3: Construct Assembly and Validation
The sgRNA is cloned into a plasmid expressing Cas9 and a selectable marker. The plasmid is validated by sequencing to confirm correct sgRNA insertion. The donor template is synthesized commercially and quality-checked.
Example:
- Use of pSpCas9(BB)-2A-GFP plasmid to enable fluorescence-based sorting of transfected cells.
Step 4: Delivery into iPSCs
Electroporation is chosen for delivery due to its efficiency in stem cells. The plasmid and donor ssODN are co-electroporated into iPSCs under optimized conditions to balance viability and editing efficiency.
Best Practice:
- Use of ROCK inhibitor post-electroporation to enhance cell survival.
- Include a non-targeting control to assess background effects.
Mind Map: Delivery Strategy
Step 5: Selection and Screening
After recovery, GFP-positive cells are sorted by flow cytometry to enrich for transfected cells. Single-cell clones are expanded and screened by PCR and Sanger sequencing to identify corrected alleles.
Example:
- PCR primers flanking the mutation site amplify a 400 bp fragment.
- Sequencing chromatograms show correction of the mutant base and presence of silent mutations.
Step 6: Validation of Edited Stem Cells
Edited clones undergo additional validation:
- Off-target analysis using targeted deep sequencing at predicted sites.
- Pluripotency marker expression assessed by immunostaining.
- Differentiation potential tested by directed differentiation into erythroid lineage.
Mind Map: Validation Workflow
Step 7: Functional Assay
To confirm functional correction, differentiated erythroid cells from edited iPSCs are analyzed for hemoglobin production. High-performance liquid chromatography (HPLC) quantifies the ratio of normal to abnormal hemoglobin.
Example:
- Edited cells show restored adult hemoglobin levels compared to unedited controls.
Summary
This example illustrates the integration of design, delivery, and validation steps in editing a disease-associated gene in stem cells. The process emphasizes careful sgRNA and donor design, efficient delivery, rigorous screening, and functional validation. Each step includes practical considerations and examples to guide successful genome editing projects in stem cells.
4. Cellular Engineering Techniques
4.1 Introduction to Cellular Engineering Concepts
Cellular engineering is the practice of modifying cells to perform specific functions or exhibit desired behaviors. It combines principles from molecular biology, genetics, and bioengineering to alter cellular components such as DNA, RNA, proteins, and metabolic pathways. The goal is to create cells that can be used for research, therapeutic applications, or industrial processes.
At its core, cellular engineering involves understanding how cells work and then applying precise interventions to change their properties. This can include introducing new genes, knocking out existing ones, or rewiring signaling pathways. The modifications can be temporary or permanent, depending on the method used and the intended application.
Mind Map: Core Components of Cellular Engineering
Cellular Engineering
├── Genetic Modification
│ ├── Gene Editing (CRISPR, TALENs, ZFNs)
│ ├── Gene Insertion
│ └── Gene Silencing (RNAi, antisense)
├── Protein Engineering
│ ├── Expression of Novel Proteins
│ ├── Fusion Proteins
│ └── Post-Translational Modifications
├── Metabolic Engineering
│ ├── Pathway Optimization
│ ├── Flux Balancing
│ └── Synthetic Pathways
└── Cellular Behavior Control
├── Signal Transduction Modulation
├── Cell Differentiation
└── Cell-Cell Communication
This map highlights the main areas where cellular engineering operates. Genetic modification is often the starting point, as it provides the blueprint for changes. Protein engineering adjusts the functional molecules inside cells. Metabolic engineering focuses on the chemical reactions cells perform, and controlling cellular behavior addresses how cells respond to their environment or interact with other cells.
Example: Engineering Immune Cells for Targeted Therapy
One practical example of cellular engineering is the development of chimeric antigen receptor T cells (CAR-T cells). Researchers modify a patient’s T cells to express a synthetic receptor that recognizes cancer cells. This involves:
- Extracting T cells from the patient.
- Using viral vectors to insert the gene encoding the chimeric receptor.
- Expanding the modified cells in culture.
- Infusing them back into the patient to target tumors.
This process demonstrates several cellular engineering principles: gene insertion, protein expression (the receptor), and functional reprogramming of cell behavior.
Mind Map: Steps in Cellular Engineering Workflow
Cellular Engineering Workflow
├── Cell Isolation
├── Genetic Modification
│ ├── Vector Design
│ ├── Delivery Method Selection
│ └── Transfection/Transduction
├── Cell Culture and Expansion
├── Functional Validation
│ ├── Molecular Assays
│ └── Phenotypic Assays
└── Application Deployment
├── Therapeutic Use
└── Research Models
This workflow outlines the typical progression from obtaining cells to applying engineered cells in real-world contexts. Each step requires careful planning and optimization. For example, delivery methods vary widely depending on cell type and modification goals, ranging from electroporation to viral transduction.
Example: Metabolic Engineering in Yeast for Biofuel Production
Another example is engineering yeast cells to produce biofuels. Scientists modify metabolic pathways to increase the yield of ethanol or other fuels. This involves:
- Identifying bottlenecks in native metabolic routes.
- Introducing genes from other organisms to create synthetic pathways.
- Knocking out competing pathways that reduce product yield.
This example shows how cellular engineering can optimize chemical production by redirecting cellular metabolism.
Mind Map: Common Techniques in Cellular Engineering
Techniques
├── Genome Editing
│ ├── CRISPR-Cas9
│ ├── TALENs
│ └── ZFNs
├── Gene Delivery
│ ├── Viral Vectors
│ ├── Electroporation
│ └── Lipid Nanoparticles
├── Protein Engineering
│ ├── Site-Directed Mutagenesis
│ └── Fusion Constructs
├── Cell Culture
│ ├── 2D Culture
│ └── 3D Culture/Organoids
└── Analytical Methods
├── Flow Cytometry
├── PCR and Sequencing
└── Microscopy
This map organizes the tools and methods commonly used in cellular engineering projects. Selecting the right combination depends on the cell type, desired modification, and downstream application.
In summary, cellular engineering is a multidisciplinary field focused on modifying cells at multiple levels to achieve specific goals. It requires a clear understanding of cellular systems, precise molecular tools, and robust validation methods. The examples and mind maps provided here illustrate the foundational concepts and practical approaches that define this area of biotechnology.
4.2 Engineering Immune Cells: CAR-T and Beyond
Engineering Immune Cells: CAR-T and Beyond
Immune cell engineering involves modifying immune cells to enhance their ability to recognize and eliminate disease targets, most notably cancer cells. The most established example is chimeric antigen receptor T-cell therapy (CAR-T), which has transformed treatment for certain blood cancers. However, the field extends beyond CAR-T, encompassing other immune cell types and engineering strategies.
CAR-T Cell Engineering
CAR-T cells are T lymphocytes genetically modified to express synthetic receptors that recognize specific antigens on target cells. These receptors combine an antigen-binding domain, usually derived from an antibody, with intracellular signaling domains that activate the T cell upon antigen engagement.
Key components of CAR design:
- Extracellular antigen recognition domain: Typically a single-chain variable fragment (scFv) that binds a tumor-associated antigen.
- Hinge and transmembrane domain: Connects the extracellular domain to the intracellular signaling components and anchors the receptor in the cell membrane.
- Intracellular signaling domains: Usually include CD3ζ for T-cell activation and co-stimulatory domains like CD28 or 4-1BB to enhance proliferation and persistence.
Mind Map: CAR-T Cell Structure
Example:
A CAR-T therapy targeting CD19, a protein expressed on B-cell malignancies, uses an scFv derived from an anti-CD19 antibody. The intracellular domain includes CD3ζ and 4-1BB, which together promote T-cell activation and survival, resulting in effective elimination of malignant B cells.
Engineering Process
- Isolation of T cells: Peripheral blood mononuclear cells are collected from the patient.
- Activation: T cells are stimulated ex vivo using agents such as anti-CD3/CD28 beads to promote proliferation.
- Gene transfer: Viral vectors (lentivirus or retrovirus) deliver the CAR construct into T cells.
- Expansion: Modified T cells are expanded to sufficient numbers.
- Infusion: Engineered CAR-T cells are infused back into the patient.
Beyond CAR-T: Other Immune Cell Engineering Strategies
While CAR-T cells have shown success in hematologic cancers, challenges remain for solid tumors and other diseases. This has led to exploration of engineering other immune cells and alternative receptor designs.
CAR-NK Cells
Natural killer (NK) cells are innate immune cells capable of killing tumor cells without prior sensitization. Engineering NK cells with CARs combines their natural cytotoxicity with antigen specificity.
Advantages:
- Lower risk of graft-versus-host disease (GVHD), enabling allogeneic (donor-derived) therapies.
- Potentially safer with reduced cytokine release syndrome.
Example:
CAR-NK cells targeting CD19 have been developed using cord blood-derived NK cells engineered with a CAR construct and IL-15 expression to enhance persistence.
TCR-Engineered T Cells
Instead of CARs, T-cell receptors (TCRs) can be engineered to recognize intracellular antigens presented by MHC molecules.
Key points:
- Broader antigen targeting, including peptides from mutated or viral proteins.
- MHC restriction requires matching patient HLA types.
Example:
T cells engineered with a TCR specific for a melanoma-associated antigen (e.g., MART-1) can recognize and kill melanoma cells presenting that peptide.
Macrophage Engineering
Macrophages can be engineered to enhance phagocytosis of cancer cells or modulate the tumor microenvironment.
Example:
Chimeric antigen receptor macrophages (CAR-M) expressing receptors that trigger engulfment upon antigen binding have been developed to target solid tumors.
Best Practices in Immune Cell Engineering
- Antigen selection: Choose antigens highly expressed on target cells but minimally on healthy tissues to reduce off-target effects.
- Receptor design: Optimize affinity and signaling domains to balance efficacy and safety.
- Delivery method: Viral vectors are standard but non-viral methods (e.g., electroporation of mRNA) can reduce insertional mutagenesis risks.
- Cell source: Autologous cells reduce rejection but allogeneic cells offer off-the-shelf availability; engineering must address immune compatibility.
- Functional testing: Assess cytotoxicity, cytokine production, and persistence in vitro and in vivo.
Mind Map: Immune Cell Engineering Strategies
Example Workflow: Engineering CAR-NK Cells
- Isolate NK cells from cord blood.
- Activate NK cells with cytokines (e.g., IL-2, IL-15).
- Transduce cells with lentiviral vector encoding anti-CD19 CAR and IL-15.
- Expand cells in culture.
- Test cytotoxicity against CD19-positive leukemia cells.
- Prepare cells for infusion.
This example highlights the integration of receptor design and cytokine support to improve NK cell persistence and function.
In summary, engineering immune cells involves selecting the right cell type, designing effective receptors, and optimizing delivery and expansion protocols. CAR-T cells remain the most clinically advanced, but alternative immune cells and receptor strategies expand the toolkit for targeting a wider range of diseases.
4.3 Stem Cell Engineering: Methods and Applications
Stem cell engineering involves modifying stem cells to alter their properties, functions, or differentiation potential. This field combines genome editing, cellular manipulation, and culture techniques to create cells tailored for research, therapy, or disease modeling. The main types of stem cells used are embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and adult stem cells such as hematopoietic stem cells (HSCs).
Methods in Stem Cell Engineering
-
Genetic Modification
- Genome Editing: Tools like CRISPR-Cas9 enable precise gene knockouts, knock-ins, or base editing in stem cells. This allows correction of mutations or insertion of reporter genes.
- Viral Transduction: Lentiviral or retroviral vectors introduce transgenes for stable expression, often used for lineage tracing or overexpression studies.
- Non-viral Methods: Electroporation or lipid-based transfection can deliver plasmids or ribonucleoprotein complexes with less risk of insertional mutagenesis.
-
Directed Differentiation
- Applying specific growth factors, small molecules, or transcription factors to guide stem cells toward desired lineages.
- Engineering transcription factor expression via inducible systems to control differentiation timing.
-
Cellular Reprogramming
- Converting somatic cells back to pluripotency (iPSCs) by introducing defined factors (e.g., OCT4, SOX2, KLF4, c-MYC).
- Direct lineage conversion or transdifferentiation bypasses pluripotency to produce specialized cells.
-
Epigenetic Modulation
- Using CRISPR-based epigenetic editors to activate or repress gene expression without altering DNA sequence.
- Modifying chromatin states to influence differentiation potential or cell identity.
Mind Map: Stem Cell Engineering Methods
Applications of Stem Cell Engineering
-
Disease Modeling: Engineered stem cells carrying patient-specific mutations allow in vitro study of disease mechanisms. For example, iPSCs derived from patients with genetic cardiomyopathies can be differentiated into cardiomyocytes to observe functional defects.
-
Drug Screening: Engineered stem cells provide platforms for testing drug efficacy and toxicity on relevant human cell types, reducing reliance on animal models.
-
Cell Replacement Therapy: Corrected or engineered stem cells can be differentiated into functional cells for transplantation. An example is generating dopamine-producing neurons from iPSCs for Parkinson’s disease treatment.
-
Gene Therapy Platforms: Stem cells engineered to express therapeutic proteins or corrected for genetic defects serve as vehicles for long-term treatment, such as HSCs modified to produce functional hemoglobin in sickle cell disease.
-
Developmental Biology Studies: Manipulating stem cells helps dissect gene function during differentiation and organogenesis.
Mind Map: Applications of Stem Cell Engineering
Concrete Examples
-
Example 1: Correcting a Mutation in iPSCs — Researchers isolated skin cells from a patient with a monogenic disorder, reprogrammed them into iPSCs, and used CRISPR-Cas9 to correct the disease-causing mutation. The corrected iPSCs were then differentiated into the affected cell type, demonstrating restored function.
-
Example 2: Engineering HSCs for Sickle Cell Disease — Hematopoietic stem cells were harvested from patients, edited ex vivo to disrupt the sickle mutation or reactivate fetal hemoglobin expression, and transplanted back, showing improved blood parameters.
-
Example 3: Directed Differentiation of ESCs into Pancreatic Beta Cells — By applying a sequence of growth factors mimicking embryonic development, ESCs were guided to become insulin-producing beta cells, which were then tested for glucose responsiveness.
Stem cell engineering requires careful optimization of editing efficiency, delivery methods, and differentiation protocols. Each step benefits from iterative testing and validation to ensure the engineered cells meet the intended functional criteria while maintaining genomic integrity.
4.4 Synthetic Biology Approaches in Cellular Engineering
Synthetic biology combines engineering principles with biology to design and construct new biological parts, devices, and systems or to redesign existing natural biological systems. In cellular engineering, synthetic biology provides tools to program cells with novel functions, enabling precise control over cellular behavior.
Key Concepts in Synthetic Biology for Cellular Engineering
- Modularity: Biological functions are broken down into discrete, interchangeable parts (e.g., promoters, ribosome binding sites, coding sequences).
- Standardization: Using well-characterized parts that behave predictably across different systems.
- Abstraction: Separating design layers (DNA sequence, parts, devices, systems) to simplify engineering.
- Design-Build-Test Cycle: Iterative process to design genetic circuits, build them, and test their function in cells.
Mind Map: Synthetic Biology Components in Cellular Engineering
Genetic Circuits: Programming Cellular Behavior
Genetic circuits are engineered networks of genes and regulatory elements that perform logical operations inside cells. They can control gene expression dynamically in response to environmental or intracellular signals.
Example: A toggle switch circuit uses two repressors that inhibit each other’s expression, enabling the cell to switch between two stable states. This can be used to control differentiation pathways or metabolic states.
Mind Map: Genetic Circuit Types
Synthetic Sensors and Actuators
Synthetic biology enables the design of sensors that detect specific molecules or signals and actuators that trigger cellular responses. For example, a synthetic sensor might detect a metabolite and activate a gene producing a therapeutic protein.
Example: Engineering a bacterial cell to sense inflammation markers and produce anti-inflammatory compounds only when needed.
Mind Map: Synthetic Sensors and Actuators
Metabolic Pathway Engineering
Synthetic biology allows redesigning metabolic pathways to optimize production of desired compounds or to introduce new biosynthetic capabilities.
Example: Engineering yeast cells to produce the antimalarial drug precursor artemisinic acid by introducing and optimizing a heterologous pathway.
Mind Map: Metabolic Engineering Strategies
Genome-Scale Engineering
Beyond single genes or pathways, synthetic biology techniques can modify entire genomes to reprogram cellular functions comprehensively.
Example: Recoding the genome of E. coli to remove all instances of a specific codon, freeing it for incorporation of non-standard amino acids.
Best Practices in Synthetic Biology for Cellular Engineering
- Characterize Parts Thoroughly: Use quantitative data to understand how parts behave in the target cell type.
- Use Modular Designs: Build circuits from interchangeable parts to facilitate troubleshooting and upgrades.
- Incorporate Controls: Design negative and positive controls to validate circuit function.
- Model Before Building: Computational models can predict circuit behavior and reduce trial-and-error.
- Iterate Design-Build-Test: Expect multiple rounds of optimization to achieve desired performance.
Practical Example: Engineering a Synthetic AND Gate in Mammalian Cells
- Design: Two input promoters responsive to different stimuli control expression of two transcription factors.
- Build: Assemble the genetic constructs using standardized parts and cloning methods.
- Test: Transfect mammalian cells and measure output reporter gene expression only when both inputs are present.
- Optimize: Adjust promoter strengths and codon usage to balance expression levels.
This approach allows cells to perform complex decision-making, useful in therapeutic contexts where multiple conditions must be met before activating a response.
Synthetic biology provides a versatile toolkit for cellular engineering, enabling precise and programmable control over cell function. By combining modular design, rigorous characterization, and iterative optimization, researchers can build sophisticated cellular systems tailored to specific applications.
4.5 Best Practices Illustrated: Engineering T Cells for Cancer Immunotherapy
Engineering T cells for cancer immunotherapy involves modifying a patient’s own immune cells to better recognize and attack cancer cells. This process requires precise genome editing, cellular manipulation, and careful validation to ensure safety and efficacy. Here, we outline best practices with clear examples and mind maps to guide the process.
Key Steps and Best Practices
Target Antigen Selection
- Choose a tumor-specific antigen expressed on cancer cells but minimally on healthy tissues.
- Example: CD19 is a common target in B-cell malignancies.
Designing the Chimeric Antigen Receptor (CAR)
- Structure includes an extracellular antigen-binding domain (usually a single-chain variable fragment, scFv), a transmembrane domain, and intracellular signaling domains.
- Best practice: Optimize scFv affinity to balance recognition and avoid off-target activation.
Gene Editing Strategy
- Use CRISPR-Cas9 or lentiviral vectors to insert CAR constructs into T cells.
- Example: CRISPR can target the TRAC locus to knock out endogenous TCR and insert CAR, reducing graft-versus-host disease risk.
T Cell Isolation and Activation
- Isolate T cells from patient blood using magnetic beads or flow cytometry.
- Activate T cells with anti-CD3/CD28 antibodies to promote proliferation and receptiveness to gene editing.
Delivery of CAR Construct
- Viral vectors (lentivirus or retrovirus) are common for stable integration.
- Non-viral methods like electroporation of mRNA or DNA plasmids can be used for transient expression.
Expansion and Quality Control
- Expand edited T cells in culture with cytokines like IL-2.
- Assess CAR expression, viability, and functionality before infusion.
Safety Testing
- Screen for off-target editing and insertional mutagenesis.
- Functional assays to confirm specificity and cytotoxicity against target cancer cells.
Mind Map: Engineering T Cells for Cancer Immunotherapy
Example: Editing T Cells to Target CD19
- Isolate T cells from patient blood using CD3 magnetic beads.
- Activate T cells with anti-CD3/CD28 beads for 48 hours.
- Design sgRNA targeting the TRAC locus for CRISPR-Cas9 editing.
- Prepare CAR construct encoding anti-CD19 scFv linked to CD28 and CD3ζ signaling domains.
- Deliver CRISPR components and CAR template via electroporation.
- Expand edited T cells in IL-2 containing media for 10 days.
- Validate editing by flow cytometry for CAR expression and TCR knockout.
- Perform cytotoxicity assay against CD19-positive leukemia cells to confirm function.
Mind Map: Example Workflow for CD19 CAR-T Cell Engineering
Additional Tips
- Optimize sgRNA design to minimize off-target effects; use computational tools and validate experimentally.
- Monitor cell viability closely after electroporation; adjust voltage and pulse length accordingly.
- Use multiple functional assays (e.g., cytokine release, killing assays) to fully characterize engineered T cells.
- Document all steps carefully to ensure reproducibility and regulatory compliance.
Through careful planning, design, and validation, engineering T cells for cancer immunotherapy can be performed effectively and safely. The integration of genome editing with cellular engineering principles is key to producing functional, specific, and clinically relevant T cell products.
5. Therapeutic Design Using Genome Editing
5.1 Principles of Therapeutic Genome Editing
Therapeutic genome editing refers to the intentional modification of DNA within living cells to treat or prevent disease. The core principle is to correct, disrupt, or insert genetic sequences in a way that restores or improves normal cellular function. This approach differs from traditional therapies by targeting the root genetic cause rather than symptoms.
Key Principles of Therapeutic Genome Editing
- Precision: The editing tool must target the intended DNA sequence with minimal off-target effects. Precision reduces unintended mutations that could cause harm.
- Efficiency: A sufficient proportion of cells must be edited to achieve a therapeutic benefit. Low editing rates may render the treatment ineffective.
- Safety: The process should avoid triggering immune responses, toxicity, or insertional mutagenesis. Safety encompasses both the editing mechanism and delivery method.
- Durability: The genetic change should persist long enough to provide lasting benefit. Temporary edits may require repeated treatments.
- Delivery: Effective transport of editing components into the target cells or tissues is critical. Delivery methods vary depending on the therapeutic context.
Below is a mind map summarizing these principles:
Precision in Detail
Precision hinges on the design of the editing system, often CRISPR-Cas9 or alternatives like base editors. Guide RNA sequences must match the target DNA exactly to avoid unintended cuts. Computational tools help predict off-target sites, but experimental validation remains essential.
Example: In treating a monogenic disorder like Duchenne muscular dystrophy, a single base change can restore the reading frame of the dystrophin gene. Designing a guide RNA that targets only the mutated exon reduces risks of off-target damage elsewhere in the genome.
Efficiency Considerations
Editing efficiency depends on the delivery method, cell type, and the editing tool itself. Some cells, like hematopoietic stem cells, are notoriously difficult to edit, requiring optimization of protocols.
Example: In sickle cell disease therapy, ex vivo editing of patient-derived stem cells achieves high efficiency by electroporating CRISPR components, followed by selection and expansion before reinfusion.
Safety Measures
Safety involves avoiding immune reactions to the editing components and preventing insertional mutagenesis, which can activate oncogenes. Transient expression systems and non-integrating vectors help mitigate these risks.
Example: Using adeno-associated virus (AAV) vectors for in vivo delivery limits integration events, reducing the risk of disrupting essential genes.
Durability of Edits
The therapeutic effect depends on how long the edited cells persist. In tissues with rapid turnover, repeated treatments or targeting stem cell populations may be necessary.
Example: For blood disorders, editing hematopoietic stem cells ensures that all progeny carry the corrected gene, providing a lifelong effect.
Delivery Strategies
Delivery is often the bottleneck in therapeutic genome editing. Viral vectors offer high efficiency but can provoke immune responses. Non-viral methods like lipid nanoparticles are safer but sometimes less efficient.
Example: Lipid nanoparticle delivery of CRISPR components to the liver has been successful in treating transthyretin amyloidosis by knocking down the mutant gene.
Integrated Mind Map of Therapeutic Genome Editing
In summary, therapeutic genome editing requires balancing multiple factors to achieve a safe and effective treatment. Each disease context demands tailored strategies that consider the genetic target, cell type, and delivery challenges. Understanding these principles helps guide experimental design and clinical development.
5.2 Designing Gene Therapies for Monogenic Disorders
Designing gene therapies for monogenic disorders requires a careful understanding of the genetic basis of the disease, the target cells or tissues, and the most suitable editing strategy. Monogenic disorders arise from mutations in a single gene, making them prime candidates for precise genome editing approaches. The goal is to correct or compensate for the defective gene to restore normal function.
Key Considerations in Designing Gene Therapies for Monogenic Disorders
- Mutation Type and Location: Different mutations (missense, nonsense, frameshift, deletions) require tailored strategies. For example, a nonsense mutation causing a premature stop codon might be addressed by base editing or exon skipping, while a large deletion might need gene addition.
- Target Cell Type: Whether the therapy targets stem cells, differentiated cells, or tissues influences delivery methods and editing approaches.
- Delivery Method: Efficient and safe delivery of editing components is critical. Viral vectors, lipid nanoparticles, or electroporation may be chosen based on the target cells.
- Editing Strategy: Options include gene disruption, correction, insertion, or regulation. The choice depends on the mutation and therapeutic goal.
- Safety and Off-Target Effects: Minimizing unintended edits and immune responses is essential.
Mind Map: Designing Gene Therapies for Monogenic Disorders
Editing Strategies Explained with Examples
-
Gene Correction: This involves repairing the specific mutation to restore the normal gene sequence. For example, in sickle cell disease, a single nucleotide mutation in the HBB gene causes abnormal hemoglobin. CRISPR-Cas9 combined with a repair template can correct this mutation in hematopoietic stem cells.
-
Gene Disruption: Sometimes, knocking out a harmful gene or regulatory element can be therapeutic. For instance, disrupting the CCR5 gene in T cells can confer resistance to HIV infection.
-
Gene Addition: When the mutation results in loss of function that cannot be corrected easily, adding a functional copy of the gene is an option. This is common in diseases like spinal muscular atrophy, where an additional copy of the SMN1 gene is introduced.
-
Base Editing: This technique allows precise conversion of one base to another without double-strand breaks. For example, converting a C•G base pair to a T•A base pair can correct certain point mutations causing diseases like Tay-Sachs.
-
Prime Editing: A newer method that can perform targeted insertions, deletions, and all 12 possible base-to-base conversions with fewer off-target effects. It can be used to correct mutations that are difficult to target with traditional CRISPR.
Example: Designing a Therapy for Cystic Fibrosis (CF)
- Mutation: The most common CF mutation is ΔF508, a three-base pair deletion in the CFTR gene.
- Strategy: Gene correction via homology-directed repair (HDR) or base editing to restore the missing phenylalanine.
- Target Cells: Airway epithelial cells, which are the primary affected tissue.
- Delivery: Viral vectors like AAV or lipid nanoparticles to deliver editing machinery to lung tissue.
- Challenges: Efficient delivery to airway cells, avoiding immune response, and ensuring long-term expression.
Mind Map: Case Study - Cystic Fibrosis Gene Therapy Design
Practical Tips for Designing Gene Therapies
- Thoroughly characterize the mutation: Knowing the exact mutation type and its effect on protein function guides the editing approach.
- Choose the right editing tool: Base editors for point mutations, prime editors for complex edits, or classic CRISPR-Cas9 for knockouts.
- Optimize delivery for the target tissue: For blood disorders, ex vivo editing of hematopoietic stem cells is common; for solid organs, in vivo delivery is more challenging.
- Plan for safety: Include off-target prediction and validation, and consider transient expression of editing components to reduce risks.
- Use relevant models: Test editing efficiency and functional correction in patient-derived cells or animal models before clinical application.
Designing gene therapies for monogenic disorders is a stepwise process that balances molecular precision, delivery challenges, and safety. Each disorder presents unique hurdles, but the core principles remain consistent: understand the mutation, pick the right editing approach, and validate thoroughly with an eye on clinical feasibility.
5.3 Ex Vivo vs In Vivo Editing Strategies
Genome editing for therapeutic purposes can broadly be categorized into two approaches: ex vivo and in vivo. Both have distinct workflows, advantages, challenges, and applications. Understanding these differences is critical for designing effective genome editing therapies.
Ex Vivo Genome Editing
Ex vivo editing involves removing cells from the patient, editing them outside the body, and then reintroducing the modified cells back into the patient. This approach is commonly used for cells that can be easily harvested and expanded, such as hematopoietic stem cells or T cells.
Workflow:
- Cell isolation from patient tissue (e.g., blood or bone marrow)
- Genome editing performed in controlled laboratory conditions
- Expansion and quality control of edited cells
- Conditioning of patient (if necessary) to accept edited cells
- Infusion of edited cells back into the patient
Advantages:
- Precise control over editing conditions, allowing optimization of efficiency and specificity
- Ability to screen and select successfully edited cells before transplantation
- Reduced risk of off-target effects in non-target tissues
- Easier to monitor and manage immune responses
Challenges:
- Requires cells that can be harvested and expanded ex vivo
- Conditioning regimens may be needed to allow engraftment
- Potential for cell damage during manipulation
Example:
Editing hematopoietic stem cells (HSCs) to correct sickle cell disease mutations. HSCs are isolated from the patient’s bone marrow or blood, edited using CRISPR-Cas9 to correct the mutation in the beta-globin gene, expanded, and then infused back. This method allows verification of editing success and purity before transplantation.
In Vivo Genome Editing
In vivo editing delivers genome editing components directly into the patient’s body, targeting cells within their natural environment. This method is suitable for tissues that are difficult to harvest or expand ex vivo, such as liver cells or muscle cells.
Workflow:
- Design of delivery system (viral vectors, lipid nanoparticles, etc.) targeting specific tissues
- Administration via injection or infusion
- Genome editing occurs within the target cells inside the body
Advantages:
- No need for cell extraction or transplantation
- Potentially simpler and less invasive
- Can target tissues inaccessible to ex vivo methods
Challenges:
- Delivery efficiency and specificity can be limiting
- Higher risk of off-target effects in non-target tissues
- Immune responses to delivery vehicles or editing components
- Limited ability to control or select edited cells post-delivery
Example:
In vivo editing of the liver to treat metabolic disorders. Delivery of CRISPR components via adeno-associated virus (AAV) vectors targets hepatocytes to disrupt or correct genes involved in disease pathways.
Mind Map: Ex Vivo Genome Editing
Mind Map: In Vivo Genome Editing
Comparative Summary
| Aspect | Ex Vivo Editing | In Vivo Editing |
|---|---|---|
| Cell Manipulation | Outside the body | Inside the body |
| Control over Editing | High (selection and expansion possible) | Limited (no selection post-delivery) |
| Delivery Challenges | Easier (direct to cells in culture) | Complex (targeting specific tissues) |
| Immune Response | Lower risk (cells can be shielded) | Higher risk (immune system exposed) |
| Suitable Cell Types | Hematopoietic, immune cells, stem cells | Liver, muscle, brain, other tissues |
| Clinical Examples | CAR-T cell therapies, sickle cell disease | Liver metabolic disorder treatments |
Choosing between ex vivo and in vivo genome editing depends on the disease target, cell accessibility, delivery feasibility, and safety considerations. Both strategies have proven effective in specific contexts and continue to be refined for broader applications.
5.4 Safety and Efficacy Assessment in Therapeutic Design
Safety and efficacy assessment is a cornerstone in therapeutic genome editing design. It ensures that the intended genetic modifications achieve the desired therapeutic effect without causing unintended harm. This section breaks down the critical components of safety and efficacy evaluation, illustrated with practical examples and mind maps to clarify complex relationships.
Understanding Safety in Therapeutic Genome Editing
Safety assessment focuses on identifying and mitigating risks associated with genome editing interventions. Key concerns include off-target effects, immune responses, insertional mutagenesis, and long-term stability of edits.
Safety Assessment Mind Map
Example: In a CRISPR-based therapy targeting a mutation in the dystrophin gene for Duchenne Muscular Dystrophy, researchers used GUIDE-seq to map potential off-target sites. They found minimal off-target activity, which was further reduced by using high-fidelity Cas9 variants. This step was crucial to ensure safety before moving to clinical trials.
Evaluating Efficacy
Efficacy assessment measures whether the genome editing achieves the therapeutic goal. This includes editing efficiency, functional restoration, and phenotypic correction.
Efficacy Assessment Mind Map
Example: For a gene therapy correcting sickle cell disease, efficacy was demonstrated by editing hematopoietic stem cells ex vivo to reactivate fetal hemoglobin production. Functional assays showed increased hemoglobin levels, and patients exhibited reduced disease symptoms post-transplant.
Integrated Safety and Efficacy Workflow
Combining safety and efficacy assessments provides a comprehensive view of therapeutic potential.
Example: In CAR-T cell therapies, preclinical studies include genomic analysis to detect unintended edits and functional assays to confirm tumor cell killing. Clinical trials monitor cytokine release syndrome as a safety endpoint while measuring tumor regression for efficacy.
Key Methods for Safety Assessment
- Off-Target Detection: Techniques like GUIDE-seq, Digenome-seq, and CIRCLE-seq identify unintended edits.
- Immunogenicity Testing: Evaluates immune responses to Cas proteins or delivery vectors.
- Genotoxicity Assays: Detect insertional mutagenesis or chromosomal rearrangements.
Key Methods for Efficacy Assessment
- Genotyping: PCR and sequencing quantify editing rates.
- Protein Assays: Western blot or ELISA measure restored protein expression.
- Functional Tests: Cell viability, differentiation, or metabolic assays assess phenotypic correction.
Practical Example: Safety and Efficacy in a CRISPR Therapy for Beta-Thalassemia
- Editing Efficiency: Achieved 70% editing in hematopoietic stem cells targeting the BCL11A enhancer.
- Off-Target Analysis: GUIDE-seq revealed no significant off-target cleavage.
- Functional Restoration: Increased fetal hemoglobin production confirmed by HPLC.
- In Vivo Safety: Mouse models showed no adverse effects or tumor formation.
- Clinical Monitoring: Patients monitored for immune reactions and hematologic parameters.
This example highlights the iterative process of balancing editing efficiency with safety to design a viable therapeutic.
Summary
Safety and efficacy assessments are intertwined processes that require multiple complementary methods. Clear, stepwise evaluation from molecular to clinical levels ensures that genome editing therapies are both effective and safe for patients.
5.5 Case Study: CRISPR-Based Therapy for Sickle Cell Disease
Sickle Cell Disease (SCD) is a genetic disorder caused by a single point mutation in the β-globin gene (HBB), leading to abnormal hemoglobin (HbS) that distorts red blood cells into a sickle shape. These misshapen cells cause blockages in blood vessels, leading to pain, organ damage, and reduced lifespan. CRISPR-based therapies aim to correct or compensate for this mutation at the genetic level.
Understanding the Genetic Target
The mutation responsible for SCD is a single nucleotide substitution (Glu6Val) in the HBB gene. The therapeutic strategies using CRISPR generally fall into two categories:
- Direct Correction: Editing the mutated HBB gene to restore the normal sequence.
- Reactivation of Fetal Hemoglobin (HbF): Disrupting regulatory elements to increase expression of the gamma-globin genes (HBG1 and HBG2), which produce HbF, a form of hemoglobin that can compensate for defective HbS.
Mind Map: Therapeutic Strategies for SCD
Step 1: Designing the CRISPR Components
For direct correction, a guide RNA (gRNA) is designed to target the mutation site in the HBB gene. A donor DNA template is provided to enable homology-directed repair (HDR), replacing the mutant base with the correct one.
For HbF reactivation, the gRNA targets an erythroid-specific enhancer region of the BCL11A gene, a transcriptional repressor of gamma-globin. Disrupting this enhancer reduces BCL11A expression, lifting repression on HbF.
Example: A commonly used gRNA targets the +58 kb erythroid enhancer of BCL11A, which is active only in red blood cell precursors, minimizing effects on other tissues.
Step 2: Delivery to Hematopoietic Stem and Progenitor Cells (HSPCs)
The therapy involves extracting HSPCs from the patient’s bone marrow or peripheral blood. CRISPR components are delivered ex vivo, typically via electroporation of ribonucleoprotein (RNP) complexes combined with donor DNA (for HDR).
Best Practice: Using RNPs reduces the time CRISPR components remain in the cell, lowering off-target risks. Electroporation parameters are optimized to balance editing efficiency and cell viability.
Step 3: Editing Efficiency and Validation
Post-editing, cells are cultured briefly to assess editing efficiency. Techniques include:
- PCR and Sanger sequencing: To confirm on-target edits.
- Next-generation sequencing (NGS): For precise quantification of editing and off-target analysis.
- Flow cytometry: To assess HbF expression levels after differentiation.
Example: In a typical protocol, editing efficiencies of 40-60% for BCL11A enhancer disruption have been reported, with corresponding increases in HbF expression.
Step 4: Conditioning and Transplantation
Edited HSPCs are infused back into the patient after myeloablative conditioning, which clears existing bone marrow cells to make space for the corrected cells.
Step 5: Clinical Outcomes and Monitoring
Successful engraftment leads to the production of red blood cells with increased HbF or corrected β-globin, reducing sickling and symptoms.
Regular monitoring includes:
- Complete blood counts
- HbF quantification
- Assessment of vaso-occlusive events
Mind Map: Workflow of CRISPR Therapy for SCD
Challenges and Solutions
-
HDR Efficiency: HDR is less efficient in HSPCs compared to NHEJ. To improve this, synchronized cell cycle manipulation or small molecules enhancing HDR are sometimes used.
-
Off-Target Effects: Careful gRNA design and transient delivery of CRISPR components help minimize unintended edits.
-
Cell Viability: Electroporation can reduce cell viability; optimizing voltage and pulse duration is critical.
-
Engraftment: Edited cells must retain stemness and engraft efficiently; culture conditions are adjusted to maintain these properties.
Concrete Example: Editing BCL11A Enhancer
A research protocol uses a chemically modified synthetic gRNA targeting the +58 kb BCL11A enhancer combined with Cas9 protein. After electroporation into mobilized peripheral blood CD34+ cells, the cells show ~50% editing efficiency. Differentiation into erythroid lineage results in a 3-4 fold increase in HbF levels compared to unedited controls.
This approach avoids the complexities of HDR and has advanced into clinical trials, showing promising safety and efficacy profiles.
Summary
CRISPR-based therapies for SCD focus on either correcting the causative mutation or reactivating fetal hemoglobin to compensate for defective adult hemoglobin. The process involves precise design of CRISPR components, efficient delivery to patient stem cells, rigorous validation, and careful clinical management. Practical examples demonstrate that disrupting the BCL11A enhancer is a viable and effective strategy with current technology.
This case study illustrates how genome editing can be translated from molecular design to therapeutic application with clear, stepwise procedures and measurable outcomes.
6. Delivery Systems for Genome Editing and Cellular Engineering
6.1 Viral Vectors: Types, Advantages, and Limitations
Viral vectors are engineered viruses used to deliver genetic material into cells. Their natural ability to infect cells makes them efficient tools for genome editing and cellular engineering. However, each viral vector type has distinct characteristics that influence its suitability for specific applications.
Common Types of Viral Vectors
-
Adenoviral Vectors (AdV)
- Derived from adenoviruses, these vectors can infect dividing and non-dividing cells.
- They do not integrate into the host genome, which reduces the risk of insertional mutagenesis.
- They typically induce strong immune responses.
-
Adeno-Associated Viral Vectors (AAV)
- Small, non-pathogenic viruses that infect dividing and non-dividing cells.
- They have limited packaging capacity (~4.7 kb).
- Mostly remain episomal but can integrate at low frequency.
- Generally elicit mild immune responses.
-
Lentiviral Vectors (LV)
- Derived from lentiviruses, a subclass of retroviruses.
- Capable of integrating into the host genome, enabling stable, long-term expression.
- Can infect dividing and non-dividing cells.
- Moderate packaging capacity (~8 kb).
-
Retroviral Vectors (RV)
- Derived from simple retroviruses.
- Require dividing cells for integration.
- Integration can cause insertional mutagenesis.
- Packaging capacity similar to lentiviruses.
Mind Map: Viral Vector Types and Key Features
Advantages of Viral Vectors
- High Transduction Efficiency: Viruses have evolved to enter cells efficiently, making them effective delivery vehicles.
- Broad Tropism: Many viral vectors can infect a wide range of cell types, including hard-to-transfect primary cells.
- Stable Expression: Integrating vectors like lentiviruses enable long-term gene expression, useful for permanent genome edits.
- Transient Expression: Non-integrating vectors such as adenoviral and AAV vectors allow temporary expression, which can be desirable in some therapeutic contexts.
Limitations and Considerations
- Immune Response: Some viral vectors, especially adenoviruses, can trigger immune reactions that reduce effectiveness and pose safety concerns.
- Packaging Size Constraints: AAV vectors have a limited capacity for genetic payloads, restricting the size of deliverable constructs.
- Insertional Mutagenesis: Integrating vectors can disrupt host genes or regulatory elements, potentially causing oncogenesis.
- Production Complexity: Manufacturing viral vectors requires specialized facilities and quality control to ensure safety and consistency.
Mind Map: Advantages vs Limitations
Examples
-
Adenoviral Vector Example: Used in ex vivo gene editing of airway epithelial cells to transiently express CRISPR-Cas9 components targeting a cystic fibrosis mutation. The non-integrating nature limits long-term effects but reduces insertional risks.
-
AAV Vector Example: Delivery of a corrective gene for retinal diseases where small payload size fits within AAV capacity, and mild immune response is critical for patient safety.
-
Lentiviral Vector Example: Engineering hematopoietic stem cells ex vivo for stable expression of a therapeutic gene in sickle cell disease. Integration ensures persistence through cell divisions.
-
Retroviral Vector Example: Historically used for gene therapy in dividing cells such as bone marrow progenitors, but concerns over insertional mutagenesis have reduced their use.
In summary, viral vectors remain a cornerstone of genome editing delivery strategies. Choosing the right vector involves balancing efficiency, safety, payload size, and the target cell type. Understanding these factors helps design experiments and therapies that meet specific goals while managing risks.
6.2 Non-Viral Delivery Methods: Electroporation, Lipid Nanoparticles, and More
Non-viral delivery methods provide alternatives to viral vectors for introducing genome editing components into cells. These methods often offer advantages in terms of safety, ease of use, and flexibility, though they come with their own challenges. This section covers key non-viral delivery techniques, focusing on electroporation, lipid nanoparticles, and other physical and chemical methods. Each method will be explained with examples and practical considerations.
Electroporation
Electroporation uses short electrical pulses to create temporary pores in the cell membrane, allowing nucleic acids, proteins, or ribonucleoprotein complexes (RNPs) to enter the cell. It is widely used for delivering CRISPR components, especially in hard-to-transfect cells like primary cells and stem cells.
How Electroporation Works
- Electrical pulses disrupt the lipid bilayer.
- Pores form transiently, permitting cargo entry.
- Membrane reseals after the pulse.
Advantages
- High delivery efficiency for many cell types.
- Can deliver DNA, RNA, or protein complexes.
- No need for viral packaging.
Limitations
- Can cause cell death if parameters are not optimized.
- Requires specialized equipment.
Example: Editing Human T Cells
In a typical protocol, human T cells are electroporated with Cas9 RNP complexes targeting a gene like PD-1. Optimizing voltage and pulse duration is critical to balance editing efficiency and viability. Post-electroporation, cells are cultured with cytokines to support recovery and expansion.
Mind Map: Electroporation
Electroporation
├── Mechanism
│ ├── Electrical pulses
│ ├── Membrane pore formation
│ └── Cargo entry
├── Cargo Types
│ ├── DNA
│ ├── RNA
│ └── Protein/RNP
├── Advantages
│ ├── High efficiency
│ └── Versatile cargo
├── Limitations
│ ├── Cell viability impact
│ └── Equipment needed
└── Applications
└── Primary cells, stem cells, immune cells
Lipid Nanoparticles (LNPs)
Lipid nanoparticles are spherical vesicles composed of lipids that encapsulate nucleic acids or proteins. They protect cargo from degradation and facilitate cellular uptake via endocytosis. LNPs have gained attention for delivering mRNA and CRISPR components in vitro and in vivo.
Composition
- Ionizable lipids: facilitate endosomal escape.
- Helper lipids: stabilize structure.
- Cholesterol: adds rigidity.
- PEG-lipids: improve circulation time.
Delivery Process
- LNPs bind to cell surface.
- Internalized by endocytosis.
- Cargo released into cytoplasm after endosomal escape.
Advantages
- Non-immunogenic compared to viral vectors.
- Scalable production.
- Suitable for systemic delivery.
Challenges
- Endosomal escape efficiency varies.
- Potential toxicity at high doses.
Example: mRNA Delivery to Liver Cells
LNPs loaded with Cas9 mRNA and guide RNA have been used to edit genes in hepatocytes. The lipid formulation is optimized to target the liver, and dosing is adjusted to minimize toxicity while achieving therapeutic editing levels.
Mind Map: Lipid Nanoparticles
Lipid Nanoparticles
├── Components
│ ├── Ionizable lipids
│ ├── Helper lipids
│ ├── Cholesterol
│ └── PEG-lipids
├── Delivery Steps
│ ├── Cell binding
│ ├── Endocytosis
│ └── Endosomal escape
├── Advantages
│ ├── Non-immunogenic
│ ├── Scalable
│ └── Systemic delivery
├── Challenges
│ ├── Variable escape
│ └── Toxicity concerns
└── Applications
└── mRNA, siRNA, CRISPR components
Other Non-Viral Methods
Chemical Transfection
Chemical reagents like lipofectamine or polyethyleneimine (PEI) form complexes with nucleic acids to facilitate cellular uptake. These methods are common in cell lines but often less effective in primary cells.
Physical Methods
- Microinjection: Direct injection of cargo into cells, useful for single-cell editing but low throughput.
- Particle Bombardment (Biolistics): DNA-coated particles are shot into cells, mostly used in plant cells.
- Sonoporation: Ultrasound waves create transient pores.
Example: Lipofectamine-Mediated CRISPR Delivery
In HEK293 cells, lipofectamine can deliver plasmids encoding Cas9 and sgRNA. Transfection efficiency and toxicity depend on reagent-to-DNA ratios and cell confluency.
Mind Map: Other Non-Viral Methods
Other Non-Viral Methods
├── Chemical
│ ├── Lipofectamine
│ └── PEI
├── Physical
│ ├── Microinjection
│ ├── Biolistics
│ └── Sonoporation
├── Advantages
│ ├── Simplicity
│ └── No viral components
├── Limitations
│ ├── Cell type specificity
│ └── Lower efficiency in some cells
└── Applications
└── Cell lines, plants, specialized cells
Summary
Non-viral delivery methods offer a toolbox for genome editing that can be tailored to the target cell type and experimental goals. Electroporation stands out for its broad applicability and efficiency, especially with RNP delivery. Lipid nanoparticles provide a promising route for in vivo applications, particularly for mRNA delivery. Chemical and physical methods fill niche roles depending on cell type and throughput needs. Choosing the right method involves balancing efficiency, cell viability, cargo type, and experimental context.
6.3 Physical Delivery Techniques: Microinjection and Biolistics
Physical delivery methods introduce genome editing components directly into cells by mechanical means rather than relying on biological vectors or chemical carriers. Two widely used physical techniques are microinjection and biolistics. Both have distinct advantages and limitations depending on the cell type, target tissue, and experimental goals.
Microinjection
Microinjection involves using a fine glass needle to physically inject nucleic acids, proteins, or ribonucleoprotein complexes into individual cells. It allows precise delivery of genome editing reagents into the cytoplasm or nucleus.
-
Procedure:
- Cells are immobilized on a microscope stage.
- A micropipette needle, pulled to a diameter of about 0.5–1 µm, is filled with the editing material.
- Under microscopic guidance, the needle penetrates the cell membrane and deposits the payload.
-
Applications:
- Commonly used in zygotes and embryos for generating genetically modified animals.
- Useful for large cells like oocytes or early embryos where other delivery methods struggle.
- Applied in cultured cells when precise control over delivery is required.
-
Advantages:
- High delivery precision and control over dosage.
- Minimal off-target delivery to neighboring cells.
- Immediate delivery without reliance on cellular uptake mechanisms.
-
Limitations:
- Labor-intensive and low throughput.
- Requires specialized equipment and skilled operators.
- Can cause mechanical damage leading to cell death if not performed carefully.
Example: Microinjection of CRISPR-Cas9 Ribonucleoproteins into Mouse Zygotes
In a typical experiment, purified Cas9 protein complexed with guide RNA is microinjected into the pronucleus of a fertilized mouse egg. This direct delivery enables efficient genome editing before the first cell division. The injected zygotes are then cultured briefly and implanted into surrogate mothers. This method yields high editing efficiency with minimal mosaicism.
Mind Map: Microinjection Workflow
Biolistics (Particle Bombardment)
Biolistics delivers DNA or other molecules by coating them onto microscopic particles (usually gold or tungsten) and shooting them into target cells or tissues using a gene gun.
-
Procedure:
- Microparticles are coated with nucleic acids or proteins.
- The particles are loaded into a gene gun device.
- A burst of pressurized gas accelerates the particles toward the target cells.
- Particles penetrate cell walls or membranes, releasing their cargo inside.
-
Applications:
- Widely used in plant cells, which have rigid cell walls that limit other delivery methods.
- Applied in some animal tissues and cultured cells where viral or chemical methods are inefficient.
- Useful for transient expression studies or stable transformation.
-
Advantages:
- Can deliver material into cells that are difficult to transfect otherwise.
- Does not rely on biological vectors, avoiding associated biosafety concerns.
- Can target tissues or intact organs directly.
-
Limitations:
- Potential for physical damage to cells and tissues.
- Delivery is somewhat random; not all cells receive particles.
- Requires optimization of particle size, velocity, and target distance.
Example: Biolistic Delivery of CRISPR Components into Plant Leaves
In a study targeting a gene in wheat leaves, gold particles coated with plasmids encoding Cas9 and guide RNA were shot into leaf tissue. After bombardment, edited cells were identified by PCR and sequencing. This method bypassed the need for tissue culture regeneration steps common in plant transformation.
Mind Map: Biolistics Process
Comparison and Integration
| Aspect | Microinjection | Biolistics |
|---|---|---|
| Target Cells | Single cells, embryos, oocytes | Plant cells, tissues, some animal cells |
| Throughput | Low (one cell at a time) | Moderate to high (many cells simultaneously) |
| Equipment | Micromanipulator, microscope | Gene gun, particle preparation tools |
| Delivery Precision | High (targeted injection) | Low (random particle distribution) |
| Cell Damage | Possible mechanical damage | Possible physical damage from particles |
| Typical Use Cases | Genetic modification of embryos, precise editing | Plant transformation, tissue-level delivery |
Both techniques are valuable for specific scenarios where chemical or viral delivery is unsuitable. Microinjection excels in precision and control, while biolistics enables delivery into challenging tissues like plants.
Summary
Physical delivery techniques like microinjection and biolistics provide direct means to introduce genome editing tools into cells. Understanding their workflows, strengths, and limitations helps in selecting the appropriate method for the biological system and experimental goals. Practical examples demonstrate their continued relevance in both research and applied biotechnology.
6.4 Optimizing Delivery Efficiency and Minimizing Toxicity
Optimizing delivery efficiency while minimizing toxicity is a critical balance in genome editing and cellular engineering. Efficient delivery ensures that the editing components reach their target cells and perform the intended modifications, while low toxicity preserves cell viability and function. This section breaks down key factors and strategies to achieve this balance, illustrated with practical examples and mind maps.
Key Factors Influencing Delivery Efficiency and Toxicity
Strategies to Optimize Delivery Efficiency
-
Choosing the Right Delivery Method for the Cell Type
- Example: Electroporation works well for T cells but can kill sensitive neurons. Lipid nanoparticles may be better for primary neurons.
-
Optimizing Cargo Format
- Using RNP complexes instead of plasmid DNA reduces the time editing components stay in the cell, lowering off-target effects and toxicity.
-
Titrating Dosage
- Start with low concentrations and gradually increase to find the sweet spot where editing efficiency plateaus but toxicity remains low.
-
Improving Delivery Vehicle Formulation
- For lipid nanoparticles, adjusting lipid composition can enhance cellular uptake and endosomal escape.
-
Preconditioning Cells
- Synchronizing cells to a particular cell cycle phase can improve editing efficiency.
Strategies to Minimize Toxicity
-
Reducing Exposure Time
- Deliver transient forms of editing components (mRNA or RNP) to limit cellular stress.
-
Using Cytoprotective Agents
- Adding antioxidants or ROCK inhibitors post-delivery can improve cell survival.
-
Optimizing Electroporation Parameters
- Adjust voltage, pulse length, and number of pulses to reduce membrane damage.
-
Avoiding Immune Activation
- Use purified components and endotoxin-free reagents.
-
Monitoring Cell Health
- Regularly assess viability and function post-delivery to catch toxicity early.
Mind Map: Balancing Efficiency and Toxicity
Example 1: Lipid Nanoparticle Delivery in Primary T Cells
Researchers aimed to deliver CRISPR-Cas9 RNPs into primary human T cells. Initial trials with high lipid nanoparticle concentrations yielded 80% editing efficiency but 40% cell death. By reducing lipid concentration by 30% and adding a ROCK inhibitor post-delivery, they maintained 75% editing efficiency while improving viability to 85%. This showed that small adjustments in formulation and post-treatment can significantly reduce toxicity without sacrificing efficiency.
Example 2: Electroporation Optimization in Neural Progenitors
Neural progenitor cells are sensitive to electroporation. A lab tested different pulse voltages and durations. High voltage pulses (1500 V) caused 60% cell death despite 70% editing efficiency. Lowering voltage to 900 V and increasing pulse number improved cell survival to 80% with a modest drop in editing to 60%. This trade-off was acceptable for downstream applications requiring viable cells.
Mind Map: Electroporation Parameter Optimization
Summary
Optimizing delivery efficiency and minimizing toxicity requires a tailored approach considering the delivery method, cell type, cargo format, and experimental conditions. Iterative testing and careful monitoring are essential. Small changes in dosage, delivery parameters, or cell treatment can shift the balance significantly. The goal is to achieve sufficient editing with minimal impact on cell health, ensuring reliable and reproducible results.
6.5 Example Protocol: Lipid Nanoparticle-Mediated Delivery in Primary Cells
Lipid nanoparticles (LNPs) have become a preferred method for delivering genome editing tools, especially in primary cells that are often difficult to transfect. This protocol outlines a step-by-step approach to prepare, characterize, and apply LNPs for delivering CRISPR-Cas9 ribonucleoproteins (RNPs) into primary human T cells. The focus is on practical details, common pitfalls, and optimization tips.
Mind Map: Key Components of LNP-Mediated Delivery
Materials
- Ionizable lipid (e.g., DLin-MC3-DMA or equivalent)
- DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine)
- Cholesterol
- PEG-lipid (e.g., DMG-PEG2000)
- CRISPR-Cas9 protein
- Synthetic sgRNA targeting gene of interest
- Ethanol (molecular biology grade)
- Citrate buffer (pH 4.0)
- Primary human T cells (isolated and cultured)
- Electroporation cuvettes (optional for comparison)
- Dynamic light scattering (DLS) instrument
- Cell culture media and supplements
Step 1: Preparation of CRISPR-Cas9 RNP Complex
- Mix Cas9 protein with sgRNA at a molar ratio of 1:1.2.
- Incubate at room temperature for 10 minutes to allow complex formation.
Example: For 10 µg Cas9, add 12 µg sgRNA in nuclease-free buffer.
Step 2: Lipid Solution Preparation
- Dissolve lipids in ethanol at the following molar ratios: ionizable lipid (50%), DSPC (10%), cholesterol (38.5%), PEG-lipid (1.5%).
- Prepare total lipid concentration at 10 mM.
Note: Ratios can be adjusted based on cell type and delivery efficiency.
Step 3: Nanoparticle Formation via Microfluidic Mixing
- Prepare aqueous phase: dilute RNP complex in 25 mM citrate buffer (pH 4.0).
- Load lipid solution and aqueous RNP solution into separate syringes.
- Use a microfluidic mixer to combine solutions at a flow rate ratio of 1:3 (lipid:aqueous).
- Collect LNP suspension immediately.
Tip: Rapid mixing promotes uniform particle size and encapsulation.
Step 4: Post-Formulation Processing
- Dialyze or buffer-exchange LNP suspension into PBS (pH 7.4) to remove ethanol and adjust pH.
- Filter through a 0.22 µm filter to sterilize.
Step 5: Characterization of LNPs
- Measure particle size and polydispersity index (PDI) using DLS; aim for 80-120 nm and PDI < 0.2.
- Determine zeta potential; near-neutral charge is typical at physiological pH.
- Assess encapsulation efficiency by quantifying unencapsulated RNP via fluorescence or gel electrophoresis.
Example: Encapsulation efficiency > 80% indicates good formulation.
Step 6: Delivery to Primary T Cells
- Count and resuspend T cells at 1 million cells/mL in complete culture medium.
- Add LNPs to cells at a dose corresponding to 1-2 µg Cas9 per million cells.
- Incubate cells with LNPs at 37°C, 5% CO2 for 24-48 hours.
Note: Optimize dosage and incubation time to balance editing efficiency and viability.
Step 7: Validation of Genome Editing
- Harvest cells post-incubation.
- Extract genomic DNA.
- Perform PCR amplification of target locus.
- Analyze editing efficiency using T7E1 assay, Sanger sequencing with ICE analysis, or next-generation sequencing.
- Assess cell viability using trypan blue exclusion or flow cytometry with viability dyes.
Example: Achieving 50-70% editing efficiency with >80% viability is a reasonable benchmark.
Mind Map: Troubleshooting and Optimization
Practical Example
In a recent experiment, primary human T cells were treated with LNPs encapsulating Cas9 RNP targeting the PD-1 gene. Using the above protocol, LNPs averaged 95 nm in diameter with a PDI of 0.15. After 48 hours, T7E1 assay showed 60% editing efficiency with 85% cell viability. Adjusting the ionizable lipid ratio from 50% to 45% improved viability to 90% but reduced editing to 55%, illustrating the trade-off between efficiency and toxicity.
This protocol balances clarity and detail to guide researchers through LNP-mediated delivery in primary cells. Each step includes considerations to adapt the method to specific experimental needs.
7. Off-Target Effects and Genome Editing Specificity
7.1 Understanding Off-Target Activity: Causes and Consequences
Genome editing tools like CRISPR-Cas9 rely on sequence-specific recognition to make precise cuts in DNA. However, the system is not perfect. Off-target activity occurs when the editing machinery binds and cuts DNA sequences similar, but not identical, to the intended target. This can lead to unintended mutations, which may affect gene function or genomic stability.
Causes of Off-Target Activity
Off-target effects arise from several factors related to the molecular mechanics of genome editing:
-
Sequence Similarity: The guide RNA (gRNA) directs Cas9 to a target sequence by base pairing. If other genomic sites share partial complementarity, Cas9 may bind and cleave these unintended sites.
-
Mismatch Tolerance: Cas9 tolerates mismatches between the gRNA and DNA, especially in certain regions of the guide sequence. Mismatches near the PAM-proximal ‘seed’ region usually reduce binding, but mismatches elsewhere might be tolerated.
-
PAM Sequence Flexibility: Cas9 requires a protospacer adjacent motif (PAM) to bind. Some Cas9 variants recognize multiple PAM sequences, increasing potential off-target sites.
-
Chromatin Accessibility: DNA regions that are more open or accessible in the chromatin structure are more likely to be bound and cut, even if the sequence match is imperfect.
-
Cas9 Concentration and Exposure Time: Higher levels of Cas9 and longer exposure increase the chance of off-target cleavage.
-
Guide RNA Design: Poorly designed gRNAs with repetitive or low-complexity sequences can increase off-target risks.
Consequences of Off-Target Activity
Unintended edits can have various outcomes depending on the location and nature of the off-target event:
-
Gene Disruption: Cutting within coding regions or regulatory elements can disrupt gene function, potentially causing loss-of-function mutations.
-
Genomic Instability: Double-strand breaks at off-target sites can lead to chromosomal rearrangements, deletions, or insertions.
-
Cellular Toxicity: Accumulation of DNA damage may trigger apoptosis or senescence.
-
Misinterpretation of Experimental Results: Off-target effects can confound phenotypic analyses, making it unclear if observed changes are due to the intended edit.
-
Safety Concerns in Therapeutics: In clinical applications, off-target mutations raise safety issues, including oncogenic risks.
Mind Map: Causes of Off-Target Activity
Mind Map: Consequences of Off-Target Activity
Examples
-
Mismatch Tolerance Example: A gRNA designed to target the sequence 5’-GAGTCCGAGCAGAAGAAGA-3’ may also bind to a site differing by one or two nucleotides, especially if mismatches are located toward the 5’ end of the guide. For instance, a site with a single mismatch at position 18 (counting from PAM-proximal end) might still be cleaved, leading to an off-target cut.
-
Chromatin Accessibility Example: In an experiment targeting a gene in human cells, off-target cuts were more frequent in euchromatic regions due to their open structure, while heterochromatic regions showed fewer off-target events despite sequence similarity.
-
Cas9 Concentration Example: Increasing the amount of Cas9 protein in a delivery system from 10 nM to 50 nM raised off-target cleavage frequency by threefold in a model cell line, illustrating the importance of dosing.
-
Therapeutic Context Example: In a clinical trial for sickle cell disease, careful screening revealed low-frequency off-target mutations in non-coding regions. While these did not disrupt known genes, the monitoring highlighted the necessity of comprehensive off-target assessment before therapy approval.
Understanding the causes and consequences of off-target activity is crucial for designing safer and more effective genome editing experiments. Careful guide RNA design, controlled delivery, and thorough validation help minimize unintended edits and their potential impacts.
7.2 Computational Prediction Tools for Off-Target Sites
Genome editing with CRISPR-Cas systems is powerful but not perfectly precise. Off-target effects—unintended edits at genomic sites similar to the target—can lead to unwanted mutations. Predicting these off-target sites computationally is a critical step in experimental design and safety assessment.
Why Computational Prediction Matters
Before running experiments, researchers use computational tools to scan the genome for sequences resembling the intended target. These tools estimate the likelihood that the CRISPR system might bind and cut at these similar sites. This helps prioritize which off-target sites to test experimentally.
Key Features of Prediction Tools
- Sequence Similarity Assessment: Most tools look for sequences with a few mismatches compared to the guide RNA (gRNA).
- Mismatch Position Weighting: Mismatches near the PAM or seed region often reduce cleavage efficiency more than those farther away.
- PAM Compatibility: Only sequences adjacent to a compatible PAM (Protospacer Adjacent Motif) are considered potential off-targets.
- Scoring Systems: Tools assign scores to potential off-targets reflecting predicted cleavage likelihood.
Mind Map: Core Components of Off-Target Prediction Tools
Common Computational Approaches
-
Alignment-Based Search: Tools scan the genome for sequences with a limited number of mismatches to the gRNA. This is often done using optimized algorithms for speed.
-
Machine Learning Models: Some tools use models trained on experimental cleavage data to predict off-target activity more accurately.
-
Thermodynamic Models: These consider binding energy and DNA accessibility to refine predictions.
Example: Predicting Off-Targets for a Sample gRNA
Suppose you have a gRNA sequence targeting the human gene HBB (beta-globin):
5'-GAGTCTGAGTCCAGGAGT-3'
A computational tool will:
- Search the human genome for sequences differing by up to 3 mismatches.
- Check if these sequences have the correct PAM (e.g., NGG for SpCas9).
- Score each potential off-target based on mismatch position.
The output might look like:
| Off-Target Sequence | Chromosome | Position | Mismatches | Score |
|---|---|---|---|---|
| GAGTCTGAGTCCAGGAGT | chr11 | 5223456 | 0 | 100 |
| GAGTCTGAGTCCAGGAGC | chr1 | 12345678 | 1 (pos 18) | 85 |
| GAGTCTGAGTTCAGGAGT | chr7 | 9876543 | 1 (pos 11) | 80 |
This table helps decide which off-targets to validate experimentally.
Mind Map: Example Workflow for Off-Target Prediction
Popular Features in Prediction Tools
- Batch Processing: Ability to analyze multiple gRNAs at once.
- Visualization: Genome browser tracks or heatmaps showing off-target distribution.
- Custom Genomes: Support for non-model organisms or modified genomes.
- Integration: Export results compatible with downstream validation tools.
Practical Example: Using a Prediction Tool in a Lab Setting
A researcher designing a CRISPR experiment targeting TP53 uses a computational tool to predict off-targets. The tool identifies 15 potential off-target sites with scores above a threshold. The researcher selects the top 5 for experimental validation using targeted deep sequencing. This approach saves time and resources by focusing on the most likely problematic sites.
Summary
Computational prediction tools are essential for anticipating off-target effects in genome editing. They combine sequence alignment, PAM recognition, and scoring algorithms to provide a ranked list of potential off-target sites. Using these predictions helps researchers design safer and more effective experiments.
7.3 Experimental Methods to Detect Off-Target Effects
Detecting off-target effects is a critical step in validating genome editing experiments. These unintended edits can compromise the safety and efficacy of applications, especially in therapeutic contexts. Several experimental approaches exist, each with its own strengths and limitations. Understanding these methods helps researchers choose the right tool for their specific needs.
Overview Mind Map
Biased Methods
Biased methods focus on a predefined set of potential off-target sites, often predicted computationally. They are useful when you have candidate sites but may miss unexpected off-targets.
Targeted Deep Sequencing
- Amplifies predicted off-target loci using PCR.
- Sequencing reads reveal mutations at these sites.
- Example: After editing a gene in HEK293 cells, researchers PCR-amplify the top 10 predicted off-target sites and sequence them to quantify indel frequencies.
GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by Sequencing)
- Introduces a short double-stranded oligodeoxynucleotide tag into double-strand breaks (DSBs).
- Tags integrate at cleavage sites, allowing their identification by sequencing.
- Example: In primary T cells, GUIDE-seq identifies both on-target and off-target cleavage sites by capturing tag integration.
SITE-seq (Selective enrichment and Identification of Tagged genomic DNA Ends by sequencing)
- Uses in vitro cleavage of purified genomic DNA by Cas9 and sequencing of cleaved ends.
- Provides a map of potential off-target sites without cellular context.
- Example: SITE-seq applied to mouse genomic DNA reveals off-target sites not predicted computationally.
Unbiased Genome-Wide Methods
These methods do not rely on prior predictions and scan the entire genome for off-target cleavage.
Digenome-seq
- Treats purified genomic DNA with Cas9 and sgRNA in vitro.
- Whole-genome sequencing identifies cleavage sites by detecting sequence patterns indicative of cuts.
- Example: Researchers use Digenome-seq to profile off-targets of a CRISPR system targeting a liver gene, finding unexpected sites.
CIRCLE-seq
- Circularizes fragmented genomic DNA.
- Cas9 cleaves circles containing target or off-target sites.
- Linearized DNA is sequenced to identify cleavage locations.
- Example: CIRCLE-seq applied to human stem cell DNA detects off-targets with high sensitivity.
DISCOVER-seq
- Monitors recruitment of DNA repair proteins (e.g., MRE11) to DSBs in living cells.
- Chromatin immunoprecipitation followed by sequencing (ChIP-seq) maps cleavage sites.
- Example: DISCOVER-seq used in edited mouse embryos identifies off-targets in a cellular context.
Functional Assays
These assays detect mismatches or indels resulting from genome editing but do not provide genome-wide information.
T7 Endonuclease I Assay
- PCR amplifies target region.
- Heteroduplex DNA formed by mixing wild-type and edited DNA is cleaved by T7 endonuclease I at mismatches.
- Cleavage products indicate editing but not precise locations.
- Example: After editing a gene in fibroblasts, T7 assay confirms presence of indels at the target site.
Surveyor Assay
- Similar to T7 assay but uses Surveyor nuclease.
- Detects mismatches with slightly different sensitivity.
- Example: Used to screen multiple clones for editing efficiency before sequencing.
Practical Example: Detecting Off-Targets in a CRISPR Experiment Targeting the BCL11A Gene
- Prediction: Use computational tools to list top 15 potential off-target sites.
- Targeted Deep Sequencing: PCR amplify these sites from edited hematopoietic stem cells and sequence to quantify indels.
- GUIDE-seq: Perform GUIDE-seq in parallel to capture unexpected off-targets.
- Validation: Confirm GUIDE-seq hits with targeted sequencing.
- Functional Assay: Use T7 endonuclease assay on select sites for quick screening.
This combined approach balances breadth and depth, ensuring comprehensive off-target detection.
Detecting off-target effects is a multi-step process often involving complementary methods. Biased approaches provide focused, sensitive detection at predicted sites, while unbiased methods reveal unexpected cleavage events. Functional assays offer quick checks but lack genome-wide scope. Choosing the right combination depends on experimental goals, cell type, and available resources.
7.4 Strategies to Minimize Off-Target Editing
Minimizing off-target editing is essential for ensuring the precision and safety of genome editing experiments. Off-target effects occur when the editing machinery, such as CRISPR-Cas9, binds and cuts DNA sequences similar but not identical to the intended target. Here are key strategies to reduce these unintended modifications.
Careful Guide RNA Design
The guide RNA (gRNA) directs the Cas nuclease to the target DNA. Designing gRNAs with high specificity is the first line of defense against off-target activity.
- Sequence Selection: Choose target sequences unique in the genome, avoiding regions with close homologs.
- Mismatch Tolerance: Avoid gRNAs with potential off-target sites differing by only one or two nucleotides, especially near the PAM-proximal seed region.
- GC Content: Aim for moderate GC content (40-60%) to balance binding strength and specificity.
Example: When editing the HBB gene, selecting a gRNA with no predicted off-targets within three mismatches reduced unintended cuts in similar globin genes.
Use of High-Fidelity Cas Variants
Engineered Cas nucleases have been developed to reduce off-target cleavage without compromising on-target efficiency.
- SpCas9-HF1: Modified to reduce nonspecific DNA interactions.
- eSpCas9: Altered to weaken binding to mismatched DNA.
- HypaCas9: Enhanced proofreading capabilities.
Example: Using SpCas9-HF1 in editing the EMX1 locus showed a 10-fold decrease in off-target cuts compared to wild-type Cas9.
Optimizing Delivery Methods and Dosage
The amount and duration of Cas9 and gRNA presence influence off-target effects.
- Transient Delivery: Use of ribonucleoprotein (RNP) complexes or mRNA reduces exposure time.
- Lower Concentrations: Minimizing nuclease and gRNA levels limits off-target activity.
- Cell Type Considerations: Some cells are more sensitive; delivery optimization is crucial.
Example: Delivering Cas9 as an RNP complex in primary T cells lowered off-target mutations compared to plasmid-based expression.
Paired Nickases and Base Editors
Using modified nucleases can improve specificity by changing the cleavage mechanism.
- Paired Nickases: Two Cas9 nickases targeting opposite DNA strands create a double-strand break only when both bind correctly.
- Base Editors: Catalyze single-base changes without double-strand breaks, reducing unintended edits.
Example: Employing paired nickases to edit the CCR5 gene reduced off-target cleavage compared to wild-type Cas9.
Incorporating Computational Prediction and Validation
Before experiments, predict potential off-target sites and validate editing outcomes.
- In Silico Prediction: Use algorithms to identify genomic sites with sequence similarity.
- Experimental Validation: Employ methods like GUIDE-seq, Digenome-seq, or targeted deep sequencing.
Example: GUIDE-seq identified unexpected off-target sites in the VEGFA gene editing, enabling redesign of the gRNA.
Temporal Control and Inducible Systems
Limiting nuclease activity to a narrow time window reduces off-target risks.
- Inducible Cas9 Systems: Cas9 activated only in presence of a small molecule or light.
- Self-Limiting Constructs: Cas9 expression controlled by feedback loops or degradation tags.
Example: Using a doxycycline-inducible Cas9 system in HEK293 cells allowed precise timing of editing, reducing off-target mutations.
Summary Mind Map
Each of these strategies addresses off-target editing from a different angle. Combining multiple approaches often yields the best results. For instance, pairing a carefully designed gRNA with a high-fidelity Cas9 delivered transiently as an RNP can significantly reduce unintended edits. Testing and validation remain crucial to confirm specificity in your particular experimental context.
7.5 Practical Example: Improving Specificity in CRISPR Editing of Neuronal Cells
Improving specificity in CRISPR editing of neuronal cells is essential because neurons are highly specialized, non-dividing cells where off-target effects can have lasting consequences. This example illustrates a step-by-step approach to enhance editing precision while maintaining efficiency.
Understanding the Challenge
Neuronal cells present unique hurdles: they are difficult to transfect, have limited DNA repair activity compared to dividing cells, and off-target edits can disrupt critical neural functions. Therefore, specificity is a priority.
Step 1: Careful Guide RNA (gRNA) Design
The first step is designing gRNAs with high specificity to the target sequence. Tools that score potential off-target sites based on sequence similarity and chromatin accessibility are useful.
- Mind Map: gRNA Design Considerations
Example: For editing the gene Syn1 in mouse cortical neurons, a gRNA targeting exon 3 was selected after screening multiple candidates for minimal predicted off-targets.
Step 2: Use of High-Fidelity Cas9 Variants
Standard Cas9 can tolerate mismatches leading to off-target cleavage. High-fidelity variants like SpCas9-HF1 or eSpCas9 have mutations reducing non-specific DNA interactions.
- Mind Map: Cas9 Variants for Specificity
Example: Using SpCas9-HF1 in primary hippocampal neurons reduced off-target editing by over 70% compared to wild-type Cas9, with comparable on-target efficiency.
Step 3: Optimizing Delivery Method
Efficient delivery minimizes the time Cas9 is active, reducing off-target events. Transient delivery methods like ribonucleoprotein (RNP) complexes are preferred over plasmids.
- Mind Map: Delivery Methods and Impact on Specificity
Example: Electroporation of Cas9 RNP complexes into cultured neurons led to precise editing with minimal off-target cleavage detected by targeted sequencing.
Step 4: Employing Paired Nickases or Base Editors
Paired nickases create single-strand breaks on opposite DNA strands, requiring two nearby gRNAs, which increases specificity. Base editors modify nucleotides without double-strand breaks, further reducing off-target risks.
- Mind Map: Alternative Editing Approaches for Specificity
Example: In a neuronal culture model, paired nickase strategy targeting Grin1 gene reduced off-target mutations by 80% compared to standard Cas9.
Step 5: Off-Target Detection and Validation
After editing, it’s critical to assess off-target effects using techniques such as GUIDE-seq, SITE-seq, or targeted deep sequencing.
- Mind Map: Off-Target Detection Methods
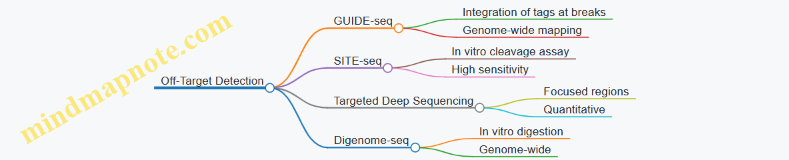
Example: GUIDE-seq applied to edited neuronal cultures revealed no detectable off-target cleavage at predicted sites when using high-fidelity Cas9 and optimized gRNAs.
Summary Table: Specificity Improvement Strategies
| Strategy | Description | Example Outcome |
|---|---|---|
| gRNA Design | Select highly specific guides | Reduced predicted off-target sites |
| High-Fidelity Cas9 | Use engineered Cas9 variants | 70% reduction in off-target cleavage |
| RNP Delivery | Transient Cas9 activity | Minimal off-target editing |
| Paired Nickases/Base Editors | Alternative editing methods | 80% off-target reduction |
| Off-Target Validation | Confirm specificity experimentally | No off-targets detected in neurons |
This example shows that improving CRISPR specificity in neuronal cells requires combining multiple strategies. Thoughtful gRNA design, choice of Cas9 variant, delivery method, and rigorous validation work together to produce precise edits with minimal unintended effects.
8. Validation and Characterization of Edited Cells
8.1 Molecular Techniques for Genotyping Edited Cells
Genotyping edited cells is a critical step in confirming the success and precision of genome editing experiments. It involves identifying the presence, absence, or nature of genetic modifications introduced by tools such as CRISPR-Cas9. This section covers the main molecular techniques used for genotyping, their principles, applications, and practical examples.
Mind Map: Molecular Genotyping Techniques
PCR-Based Methods
PCR (Polymerase Chain Reaction) is the backbone of genotyping. It amplifies specific DNA regions to detect edits.
-
Conventional PCR: Amplifies the target locus to check for insertions or deletions (indels) by size differences on a gel. For example, a 5 bp deletion will produce a slightly smaller band.
-
Quantitative PCR (qPCR): Measures DNA quantity in real-time, useful for detecting copy number changes or allele frequency shifts.
-
Digital Droplet PCR (ddPCR): Partitions the PCR reaction into thousands of droplets, allowing absolute quantification of edited versus unedited alleles with high sensitivity.
Example: After editing a gene in HEK293 cells, conventional PCR followed by gel electrophoresis showed a band shift consistent with a 10 bp deletion. ddPCR further quantified that 65% of alleles carried the edit.
Restriction Fragment Length Polymorphism (RFLP)
RFLP exploits changes in restriction enzyme recognition sites caused by edits. If a CRISPR edit disrupts a restriction site, digestion patterns will differ.
Example: Editing a target site that contains an EcoRI site. Post-edit PCR product digestion with EcoRI shows undigested bands in edited cells, confirming loss of the site.
Sanger Sequencing
Sanger sequencing remains a gold standard for precise sequence validation. PCR products are sequenced to identify exact nucleotide changes.
-
Useful for small edits or verifying single nucleotide changes.
-
Mixed sequences from heterogeneous cell populations can be deconvoluted using software or by cloning PCR products before sequencing.
Example: Sequencing a PCR amplicon from edited stem cells revealed a 2 bp insertion at the target site, confirming the intended frameshift mutation.
Next-Generation Sequencing (NGS)
NGS provides deep, high-throughput sequencing of edited loci, enabling detection of rare edits and off-target events.
-
Allows quantification of editing efficiency across thousands of reads.
-
Suitable for complex edits or pooled samples.
Example: NGS of a CRISPR-edited T cell population showed 78% on-target editing and identified low-frequency off-target mutations.
T7 Endonuclease I (T7E1) Assay
T7E1 recognizes and cleaves mismatched DNA heteroduplexes formed when edited and unedited DNA strands anneal.
-
PCR amplifies the target region.
-
Denaturation and re-annealing create heteroduplexes.
-
T7E1 digestion followed by gel electrophoresis reveals cleavage products indicating indels.
Example: After editing a gene in neuronal cells, T7E1 assay showed cleavage bands corresponding to a 30% editing efficiency.
High-Resolution Melt Analysis (HRMA)
HRMA detects sequence variations by measuring melting temperature differences of PCR amplicons.
-
Sensitive to small indels and single nucleotide polymorphisms.
-
Fast and cost-effective screening method.
Example: HRMA distinguished edited clones from wild-type by a characteristic melt curve shift, guiding selection for sequencing.
Allele-Specific PCR
This method uses primers designed to selectively amplify edited or wild-type alleles based on sequence differences.
-
Useful for detecting known point mutations.
-
Requires careful primer design to ensure specificity.
Example: Detecting a single base substitution introduced by base editing in a liver cell line using allele-specific primers.
Summary Table of Techniques
| Technique | Detects | Sensitivity | Throughput | Notes |
|---|---|---|---|---|
| Conventional PCR | Indels by size | Moderate | Low | Simple, initial screening |
| qPCR | Copy number, allele freq | High | Medium | Quantitative |
| ddPCR | Allele frequency | Very high | Low-Medium | Absolute quantification |
| RFLP | Site disruptions | Moderate | Low | Requires restriction site |
| Sanger Sequencing | Exact sequence | High (single clones) | Low | Gold standard for small edits |
| NGS | Comprehensive sequence | Very high | High | Detects rare edits, off-target |
| T7E1 Assay | Indels (mismatches) | Moderate | Low | Quick screening |
| HRMA | Small sequence changes | Moderate-High | Medium | Cost-effective screening |
| Allele-Specific PCR | Known point mutations | High | Low | Primer design critical |
Genotyping edited cells often involves combining multiple techniques. For example, initial screening with T7E1 or HRMA can identify edited clones, followed by Sanger sequencing for precise characterization. In populations with mixed edits, NGS or ddPCR provides detailed quantification.
By selecting appropriate methods based on the edit type, sample complexity, and throughput needs, researchers can efficiently confirm genome editing outcomes.
8.2 Functional Assays to Confirm Phenotypic Changes
Functional assays are essential to confirm that genome editing has produced the intended phenotypic changes in cells. These assays go beyond genotyping to assess whether the edited gene affects cell behavior, protein function, or physiological processes as expected. Choosing the right functional assay depends on the gene’s role, the cell type, and the desired outcome.
Types of Functional Assays
Functional assays can be broadly categorized based on what aspect of cell function they evaluate:
- Protein Expression and Activity
- Cellular Phenotype and Morphology
- Metabolic and Signaling Pathways
- Cell Viability and Proliferation
- Functional Output Specific to Cell Type
Below is a mind map summarizing these categories:
Protein Expression and Activity
Confirming that the edited gene produces or affects the protein of interest is often the first step. Western blotting detects protein presence and size, revealing if the edit causes truncations or loss. ELISA quantifies secreted proteins, useful for cytokines or hormones. Enzyme activity assays measure functional output directly, such as kinase activity or metabolic enzyme function.
Example: After knocking out a gene encoding a metabolic enzyme, an enzyme activity assay can confirm loss of function, while Western blot ensures the protein is absent or truncated.
Cellular Phenotype and Morphology
Changes in cell shape, size, or surface markers can indicate phenotypic shifts. Microscopy allows visualization of morphology changes or subcellular localization of proteins. Flow cytometry quantifies expression of surface markers or intracellular proteins across thousands of cells, providing population-level data.
Example: Editing a gene involved in immune cell differentiation can be validated by flow cytometry measuring changes in CD markers characteristic of specific cell types.
Metabolic and Signaling Pathways
Reporter gene assays use constructs where a reporter (like luciferase or GFP) is driven by a pathway-responsive promoter, indicating pathway activation or repression. Phosphorylation status of signaling proteins can be assessed by phospho-specific antibodies. Metabolite quantification measures changes in cellular metabolites reflecting pathway alterations.
Example: To verify disruption of a signaling pathway, a luciferase reporter under control of a responsive element can show reduced activity in edited cells.
Cell Viability and Proliferation
Editing may affect cell growth or survival. MTT or XTT assays assess metabolic activity as a proxy for viability. Direct cell counting or proliferation markers (like Ki-67 staining) provide complementary data. Apoptosis assays detect programmed cell death induced by editing.
Example: Editing a gene suspected to regulate cell cycle can be tested by measuring proliferation rates and apoptosis markers.
Functional Output Specific to Cell Type
Some assays are tailored to the specialized function of the cell. Neurons can be tested by electrophysiology to measure action potentials. Cardiomyocytes’ contractility can be quantified by video microscopy or impedance measurements. Endocrine cells’ hormone secretion can be measured by ELISA.
Example: After editing a gene in cardiomyocytes, contractility assays confirm whether the cells maintain normal beating patterns.
Integrating Functional Assays
Often, multiple assays are combined to build a comprehensive picture. For example, editing a gene in stem cells might require confirming protein loss (Western blot), changes in differentiation markers (flow cytometry), and functional differentiation capacity (cell-type specific assays).
Summary Mind Map of an Example Workflow
Functional assays provide the biological context that genotyping alone cannot. They confirm whether genome editing produces meaningful changes in cell behavior or function, which is critical for both research and therapeutic applications.
8.3 Single-Cell Analysis in Genome Editing Validation
Single-cell analysis has become an essential tool for validating genome editing outcomes. Unlike bulk assays that average signals across many cells, single-cell methods reveal the heterogeneity within edited populations. This is crucial because genome editing often produces a mix of edited, partially edited, and unedited cells, as well as varying off-target effects and mosaicism.
Why Single-Cell Analysis?
- Detects editing efficiency at the individual cell level.
- Identifies mosaicism and clonal variation.
- Links genotype to phenotype within the same cell.
- Enables characterization of rare edited cells that might be missed in bulk analysis.
Key Techniques in Single-Cell Analysis for Genome Editing
- Single-cell PCR and sequencing: Amplifies target loci from individual cells to determine precise edits.
- Single-cell RNA sequencing (scRNA-seq): Measures transcriptomic changes post-editing, revealing functional consequences.
- Flow cytometry and FACS: Sorts cells based on surface markers or fluorescent reporters linked to editing events.
- Single-cell ATAC-seq: Profiles chromatin accessibility changes after editing.
Mind Map: Single-Cell Analysis Workflow
Example 1: Single-Cell Genotyping After CRISPR Knockout
A researcher targets a gene in a heterogeneous cell line using CRISPR-Cas9. Bulk PCR suggests 70% editing efficiency, but single-cell PCR reveals three subpopulations: 50% with biallelic knockouts, 20% with monoallelic edits, and 30% unedited. This granularity informs downstream experiments, such as selecting clones for further study.
Example 2: Linking Genotype to Phenotype Using scRNA-seq
After editing a transcription factor gene, scRNA-seq identifies cells with successful edits by detecting indels in the target transcript. Comparing gene expression profiles between edited and unedited cells reveals downstream pathways affected by the knockout, clarifying the gene’s role.
Best Practices
- Maintain cell viability: Gentle dissociation and processing reduce stress-induced artifacts.
- Use appropriate controls: Include unedited cells and mock-treated samples for baseline comparisons.
- Validate single-cell isolation: Confirm that wells or droplets contain single cells to avoid mixed signals.
- Combine modalities: Integrate DNA, RNA, and protein data for a comprehensive view.
- Careful data interpretation: Account for allelic dropout and amplification biases common in single-cell assays.
Mind Map: Challenges and Solutions in Single-Cell Genome Editing Validation
Single-cell analysis adds a layer of precision to genome editing validation. It helps researchers understand not just if editing occurred, but how it varies cell-to-cell and what functional consequences arise. This clarity is essential when developing therapies or studying gene function in complex tissues.
8.4 Best Practices in Data Interpretation and Reporting
Interpreting and reporting data from genome editing experiments is a critical step that shapes conclusions and downstream decisions. Clear, accurate, and transparent communication ensures reproducibility and trust. Here are key practices to keep in mind, illustrated with examples and mind maps.
Contextualize Your Data
Always interpret results within the framework of your experimental design and biological system. Raw numbers or percentages alone rarely tell the full story.
- Example: A 30% editing efficiency in a difficult-to-transfect primary T cell population might be excellent, whereas the same number in a robust cell line could indicate suboptimal conditions.
Mind Map: Contextualizing Data
Use Appropriate Controls
Controls are your baseline for comparison and help distinguish true editing events from noise or artifacts.
- Negative controls (e.g., cells treated without guide RNA) reveal background mutations or off-target effects.
- Positive controls (e.g., known efficient sgRNA) validate your system’s responsiveness.
Example: If a negative control shows 5% indels, an observed 7% in your test sample may not be significant.
Mind Map: Controls in Genome Editing
Quantify Editing Efficiency Clearly
Report editing efficiency using standardized metrics such as:
- Indel frequency (percentage of alleles with insertions/deletions)
- HDR (homology-directed repair) rates if applicable
- Allele-specific editing when relevant
Always specify the method used (e.g., TIDE analysis, deep sequencing) and the sample size.
Example: “TIDE analysis indicated 45% indel formation at the target locus in HEK293 cells (n=3 biological replicates).”
Address Off-Target Effects Transparently
Include off-target analysis results, even if negative. Describe the methods used (computational prediction, GUIDE-seq, etc.) and any observed off-target mutations.
Example: “No off-target mutations were detected at the top five predicted sites by deep sequencing.”
Present Data Visually and Accessibly
Use graphs, tables, and annotated sequence alignments to make data easier to understand.
- Bar charts for editing efficiencies
- Sequence chromatograms or alignments showing mutations
- Flow cytometry plots for phenotypic changes
Example: A bar graph comparing editing efficiencies across different sgRNAs with error bars representing standard deviation.
Mind Map: Effective Data Presentation
Include Statistical Analysis
Apply appropriate statistical tests to support conclusions. Report p-values, confidence intervals, and number of replicates.
Example: “Editing efficiency was significantly higher with sgRNA1 compared to sgRNA2 (p=0.01, Student’s t-test, n=4).”
Discuss Limitations and Variability
No experiment is perfect. Acknowledge sources of variability such as batch effects, cell heterogeneity, or technical noise.
Example: “Variability in editing efficiency across replicates may reflect differences in transfection efficiency.”
Maintain Reproducibility and Transparency
Provide detailed methods, reagent information, and raw data when possible. This allows others to replicate and validate findings.
Example: “Guide RNA sequences, plasmid maps, and raw sequencing data are included in supplementary materials.”
Use Clear and Precise Language
Avoid ambiguous terms. Instead of “high editing,” specify exact percentages and conditions.
Example: Prefer “Editing efficiency reached 60% in Jurkat cells after 48 hours” over “Editing was high.”
Integrate Multiple Data Types
Combine molecular data (e.g., sequencing) with phenotypic assays (e.g., protein expression, cell behavior) for a comprehensive picture.
Example: “Sequencing confirmed knockout of gene X, which correlated with a 40% reduction in protein Y levels measured by Western blot.”
Mind Map: Integrated Data Interpretation
Following these practices helps produce clear, reliable, and meaningful reports that advance understanding and application of genome editing. Clear data interpretation is not just about numbers; it’s about telling the right story with the evidence you have.
8.5 Case Study: Validating Gene Knockout in iPSC-Derived Cardiomyocytes
Validating a gene knockout in induced pluripotent stem cell (iPSC)-derived cardiomyocytes involves a multi-step process that combines molecular, cellular, and functional assays. The goal is to confirm that the targeted gene is effectively disrupted and that this disruption produces the expected cellular phenotype without unintended effects.
Step 1: Confirming Gene Editing at the DNA Level
The first step is to verify that the CRISPR-Cas9 system successfully introduced mutations at the target locus. This typically involves:
- PCR Amplification: Amplify the genomic region surrounding the target site.
- Sanger Sequencing: Sequence the PCR product to identify insertions or deletions (indels).
- TIDE Analysis: Use Tracking of Indels by DEcomposition (TIDE) to quantify editing efficiency.
Example: After editing the MYH7 gene, PCR primers flanking exon 3 are designed. Sequencing reveals a 5 bp deletion causing a frameshift.
Mind Map: DNA-Level Validation
Step 2: Assessing mRNA Expression
Next, measure the transcript levels of the target gene to check if the knockout leads to mRNA degradation (e.g., via nonsense-mediated decay):
- qRT-PCR: Quantify mRNA levels relative to housekeeping genes.
- RNA-Seq (optional): Broader transcriptomic changes.
Example: qRT-PCR shows MYH7 mRNA reduced to 10% of wild-type levels, indicating successful knockout.
Mind Map: RNA-Level Validation
Step 3: Protein-Level Confirmation
Protein analysis confirms whether the knockout abolishes protein expression:
- Western Blotting: Detect protein presence and size.
- Immunocytochemistry (ICC): Visualize protein localization in cardiomyocytes.
Example: Western blot shows absence of MYH7 protein band in edited cells; ICC confirms loss of sarcomeric staining.
Mind Map: Protein-Level Validation
Step 4: Functional Characterization of Edited Cardiomyocytes
Since cardiomyocytes have distinct electrophysiological and contractile properties, functional assays help determine the biological impact of the knockout:
- Calcium Imaging: Measures intracellular calcium transients during contraction.
- Patch-Clamp Electrophysiology: Records action potentials and ion channel activity.
- Contractility Assays: Quantify beating frequency and force.
Example: MYH7 knockout cells display reduced contraction amplitude and altered calcium transient kinetics.
Mind Map: Functional Validation
Step 5: Off-Target and Clonal Variation Assessment
To ensure specificity and reproducibility:
- Off-Target Analysis: PCR and sequencing of predicted off-target sites.
- Clonal Expansion: Isolate multiple clones to confirm consistent knockout effects.
Example: Sequencing of top 5 predicted off-target loci shows no mutations; three independent clones exhibit similar phenotypes.
Mind Map: Specificity and Reproducibility
Summary Table of Validation Steps and Techniques
| Validation Level | Technique(s) | Purpose | Example Result |
|---|---|---|---|
| DNA | PCR, Sanger Sequencing, TIDE | Confirm indels at target site | 5 bp deletion causing frameshift |
| RNA | qRT-PCR | Measure transcript reduction | 90% reduction in MYH7 mRNA |
| Protein | Western Blot, ICC | Confirm protein loss | Absence of MYH7 protein band |
| Functional | Calcium Imaging, Patch Clamp | Assess cardiomyocyte function | Reduced contraction amplitude |
| Specificity | Off-target sequencing | Check for unintended edits | No off-target mutations detected |
This structured approach ensures that the gene knockout is not only present at the genetic level but also translates into the expected molecular and functional changes in iPSC-derived cardiomyocytes. Each step builds confidence in the editing outcome and provides a comprehensive validation framework.
9. Ethical and Regulatory Considerations in Genome Editing
9.1 Ethical Principles in Genome Editing Research
Ethical principles in genome editing research serve as a compass to guide scientists, clinicians, and policymakers through complex decisions. These principles help ensure that research respects human dignity, promotes fairness, and minimizes harm. Here, we break down the core ethical considerations with clear examples and mind maps to clarify their relationships.
Respect for Autonomy
At the heart of ethical research is respect for individual autonomy—the right of people to make informed decisions about their own bodies and genetic information. This means obtaining informed consent before any genome editing intervention, especially in clinical settings.
Example: Before editing a patient’s cells to treat a genetic disorder, researchers must clearly explain the procedure, risks, benefits, and alternatives. The patient’s voluntary agreement is essential.
Beneficence and Non-Maleficence
Beneficence requires that genome editing research aims to do good—improving health or knowledge—while non-maleficence insists on avoiding harm. These principles demand careful risk-benefit analysis.
Example: Editing embryos to prevent a severe genetic disease might offer significant benefits, but if off-target effects could cause unintended harm, the risks may outweigh the benefits.
Justice
Justice in genome editing means fair access to benefits and burdens. It addresses who gets to use these technologies and who might be excluded or disproportionately affected.
Example: If a gene therapy is expensive, only wealthy patients might access it, raising concerns about equity. Researchers and policymakers must consider how to make therapies accessible and avoid exacerbating health disparities.
Transparency and Accountability
Researchers must be transparent about their methods, intentions, and findings. Accountability means taking responsibility for outcomes, including unintended consequences.
Example: Publishing both successful and unsuccessful genome editing experiments helps the scientific community learn and prevents repetition of errors.
Respect for Future Generations
Genome editing, especially germline editing, affects not only current individuals but also their descendants. Ethical research considers long-term impacts and the rights of future people.
Example: Editing the germline to eliminate a hereditary disease must weigh the potential benefits against unknown effects on future generations.
Cultural Sensitivity and Social Context
Ethical genome editing recognizes diverse cultural values and social norms. What is acceptable in one community may not be in another.
Example: Some cultures may have specific views on modifying human embryos, requiring researchers to engage respectfully and adapt approaches accordingly.
Summary Mind Map
Each principle interacts with others. For instance, ensuring justice may require transparency about costs and access, while respect for autonomy depends on clear communication and cultural sensitivity. Ethical genome editing research is a balancing act, requiring ongoing reflection and dialogue among all stakeholders.
9.2 Regulatory Frameworks Governing Therapeutic Genome Editing
Regulatory frameworks for therapeutic genome editing are designed to ensure that these powerful technologies are developed and applied safely, ethically, and effectively. These frameworks vary by country but share common principles such as risk assessment, clinical trial oversight, manufacturing standards, and post-market surveillance.
Overview of Regulatory Bodies and Their Roles
- FDA (U.S. Food and Drug Administration): Oversees clinical trials and approval of gene therapies, including genome editing products.
- EMA (European Medicines Agency): Regulates advanced therapy medicinal products (ATMPs) in the European Union.
- PMDA (Pharmaceuticals and Medical Devices Agency, Japan): Reviews gene therapy products and ensures compliance with safety standards.
- Other National Agencies: Many countries have their own regulatory bodies that align with international standards but may have specific local requirements.
Key Regulatory Considerations
- Preclinical Data Requirements: Demonstrating safety and efficacy in relevant models before human trials.
- Clinical Trial Authorization: Submission of detailed protocols outlining patient selection, dosing, monitoring, and endpoints.
- Manufacturing Controls: Ensuring consistency, purity, and potency of gene editing products under Good Manufacturing Practice (GMP).
- Long-Term Follow-Up: Monitoring patients for delayed adverse effects or unintended consequences.
Mind Map: Regulatory Framework Components
Example: FDA’s Approach to CRISPR-Based Therapies
The FDA requires an Investigational New Drug (IND) application before clinical trials. This includes detailed information about the genome editing system, delivery method, target cells, and potential risks. For example, a CRISPR therapy targeting sickle cell disease must provide data on off-target effects, immune responses, and manufacturing consistency. The FDA also mandates long-term follow-up protocols to monitor for insertional mutagenesis or other delayed effects.
Mind Map: IND Application Components
International Harmonization Efforts
While regulatory agencies operate independently, there is ongoing coordination through organizations like the International Council for Harmonisation (ICH). This helps align standards for quality, safety, and efficacy, reducing duplication and facilitating global development.
Example: EMA’s Regulation of Advanced Therapy Medicinal Products (ATMPs)
The EMA classifies genome editing therapies under ATMPs, which require centralized authorization. Developers submit a Marketing Authorization Application (MAA) including clinical data, manufacturing details, and risk management plans. The EMA emphasizes traceability and pharmacovigilance, ensuring that any adverse events are promptly identified and addressed.
Mind Map: EMA ATMP Regulatory Pathway
Challenges in Regulatory Oversight
- Complexity of Genome Editing Products: Variability in editing tools and delivery methods complicates standardization.
- Off-Target Effects: Regulators require robust methods to detect and mitigate unintended edits.
- Ethical Concerns: Germline editing is heavily restricted or prohibited in many jurisdictions.
- Manufacturing Scalability: Ensuring consistent quality at scale remains a hurdle.
Best Practices for Navigating Regulatory Frameworks
- Engage early with regulatory agencies to clarify requirements.
- Develop comprehensive preclinical packages addressing safety and specificity.
- Implement rigorous quality control and documentation in manufacturing.
- Plan for long-term patient monitoring and transparent reporting.
Mind Map: Best Practices for Regulatory Compliance
In summary, regulatory frameworks for therapeutic genome editing balance innovation with patient safety. Understanding the specific requirements of each jurisdiction and integrating best practices into development plans is essential for successful translation from bench to bedside.
9.3 Informed Consent and Patient Safety
Informed consent and patient safety are cornerstones of ethical clinical practice, especially in genome editing therapies where the risks and benefits can be complex and novel. Informed consent is not just a form; it is a process of communication that ensures patients understand what they are agreeing to, including the nature of the intervention, potential risks, benefits, and alternatives.
Key Elements of Informed Consent
- Disclosure: Providing clear, accurate information about the procedure, including the genome editing technique, expected outcomes, and possible side effects.
- Comprehension: Confirming that the patient understands the information, which may require tailored explanations or educational materials.
- Voluntariness: Ensuring the patient’s decision is made freely, without coercion or undue influence.
- Competence: Assessing whether the patient has the capacity to make informed decisions.
- Consent: Obtaining explicit agreement, usually documented, before proceeding.
Mind Map: Informed Consent Process
Patient Safety Considerations
Patient safety in genome editing involves minimizing harm throughout the treatment lifecycle. This includes careful patient selection, monitoring for adverse effects, and readiness to manage complications.
- Risk Assessment: Identifying potential off-target effects, immune responses, or unintended genetic changes.
- Monitoring: Regular clinical and molecular follow-up to detect and address side effects early.
- Emergency Preparedness: Having protocols in place for adverse events.
- Transparency: Informing patients about uncertainties and unknowns inherent in new therapies.
Mind Map: Patient Safety in Genome Editing
Example: Informed Consent in a CRISPR Clinical Trial
Imagine a clinical trial testing CRISPR-based therapy for a genetic blood disorder. The informed consent process includes explaining how CRISPR edits the faulty gene, potential risks like unintended edits or immune reactions, and alternative treatments such as bone marrow transplant. The patient is given time to ask questions and is provided with simple diagrams illustrating the editing process. The clinical team confirms understanding by asking the patient to explain the procedure back in their own words. Consent is documented, but the conversation continues throughout the trial to update the patient on new findings or risks.
Example: Patient Safety Protocols
In the same trial, patient safety protocols include pre-treatment screening for immune markers, frequent blood tests post-treatment to detect off-target edits, and a 24/7 hotline for reporting symptoms. If unexpected side effects arise, the team has a predefined plan to halt treatment and provide supportive care. Patients are informed about these safety measures during consent, reinforcing trust and transparency.
Integrating Informed Consent and Safety
The process is iterative: informed consent is revisited as new data emerges, and patient safety monitoring informs ongoing communication. This integration ensures patients remain partners in their care, aware of evolving risks and benefits.
Mind Map: Integration of Informed Consent and Patient Safety
In summary, informed consent and patient safety in genome editing require clear communication, thorough understanding, and ongoing vigilance. These elements protect patients and support ethical clinical practice.
9.4 Case Examples: Navigating Regulatory Approval for Gene Therapies
Navigating regulatory approval for gene therapies involves a structured process designed to ensure safety, efficacy, and ethical compliance. Each regulatory agency has its own framework, but common themes emerge across jurisdictions. Understanding these frameworks through concrete examples clarifies the path from laboratory innovation to clinical application.
Regulatory Approval Mind Map
Case Example 1: FDA Approval of a CAR-T Cell Therapy
In 2017, the FDA approved the first CAR-T cell therapy for certain types of lymphoma. This approval process highlights several key regulatory steps:
- Preclinical Data: Extensive in vitro and animal studies demonstrated the therapy’s ability to target cancer cells without unacceptable toxicity.
- Clinical Trials: The therapy underwent a phased clinical trial process, starting with a small group to assess safety and dosage, then expanding to larger groups to confirm efficacy and monitor side effects.
- Manufacturing Controls: The therapy’s manufacturing process was validated under Good Manufacturing Practice (GMP) standards to ensure consistency and safety.
- Regulatory Submission: The company submitted a Biologics License Application (BLA) including all preclinical, clinical, and manufacturing data.
- Post-Approval Monitoring: The FDA required long-term follow-up to monitor for delayed adverse effects, given the novel nature of the therapy.
This example shows how regulatory bodies balance innovation with patient safety through rigorous data requirements and ongoing oversight.
Case Example 2: EMA Review of a Gene Therapy for Inherited Retinal Disease
The European Medicines Agency (EMA) approved a gene therapy targeting a rare inherited retinal disease. Key points from this process include:
- Orphan Drug Designation: The therapy qualified for special status due to the rarity of the condition, which provided regulatory incentives.
- Conditional Marketing Authorization: Given the limited patient population, the EMA granted conditional approval based on promising early-phase clinical data, with commitments to provide further evidence post-approval.
- Risk Management Plan: A detailed plan was required to monitor potential risks, including immune responses and long-term effects.
- Manufacturing Oversight: The EMA placed emphasis on the control of viral vector production, ensuring purity and potency.
This case illustrates how regulatory agencies adapt their frameworks to accommodate therapies for rare diseases while maintaining rigorous standards.
Regulatory Submission Process Mind Map
Practical Considerations
- Early Engagement: Interacting with regulators early in development helps clarify expectations and avoid costly delays.
- Data Quality: High-quality, reproducible data across preclinical and clinical studies is essential.
- Manufacturing Consistency: Gene therapies require tightly controlled production processes to ensure product integrity.
- Ethical Compliance: Informed consent, patient privacy, and equitable access are scrutinized throughout the approval process.
Summary
Regulatory approval for gene therapies is a multi-step process involving preclinical validation, phased clinical trials, manufacturing oversight, and post-market surveillance. Case examples from the FDA and EMA show how agencies apply these principles in practice, balancing innovation with patient safety. Clear communication, robust data, and adherence to quality standards are critical to successfully navigating this pathway.
9.5 Best Practices for Compliance and Responsible Research Conduct
Compliance and responsible conduct in genome editing research are essential to maintain scientific integrity, protect human subjects, and ensure public trust. This section outlines practical steps and examples to help researchers navigate the complex regulatory and ethical landscape.
Understanding Regulatory Requirements
Before initiating any genome editing project, familiarize yourself with the relevant local, national, and international regulations. These often include guidelines on:
- Human subject protection
- Biosafety and biosecurity
- Data privacy
- Intellectual property
Example: When working with human cells, obtaining Institutional Review Board (IRB) approval is mandatory. This process involves submitting detailed protocols that explain the purpose, methods, risks, and benefits of the research.
Informed Consent
Obtaining clear and thorough informed consent is a cornerstone of ethical research. Consent documents should:
- Use plain language avoiding jargon
- Explain the genome editing process and potential risks
- Clarify the scope of data use and sharing
- Offer the option to withdraw at any time
Example: In a study editing patient-derived stem cells, the consent form included a section explaining off-target risks and how the data would be anonymized.
Data Management and Privacy
Responsible research includes secure handling of sensitive genetic data. Best practices involve:
- Encrypting data storage
- Limiting access to authorized personnel
- Anonymizing samples and datasets
- Complying with data protection laws like GDPR or HIPAA
Example: A lab working on CRISPR therapies implemented a tiered access system where only the principal investigator and data manager could access identifiable patient information.
Transparency and Reporting
Accurate and transparent reporting of methods, results, and adverse events helps the scientific community and regulators assess the validity and safety of genome editing work. This includes:
- Documenting all experimental steps
- Reporting off-target effects and unexpected outcomes
- Sharing protocols and reagents when possible
Example: A research group published a detailed account of their CRISPR editing efficiency and off-target analysis in a peer-reviewed journal, including negative results.
Conflict of Interest Management
Disclose any financial or personal interests that could influence the research. This prevents bias and maintains credibility.
Example: A researcher developing a CRISPR-based therapy disclosed ownership in a startup company during grant applications and publications.
Continuous Training and Education
Regular training on ethical standards, biosafety, and regulatory updates ensures the research team stays compliant.
Example: A lab holds quarterly workshops on responsible conduct of research and updates on genome editing regulations.
Mind Map: Key Areas of Compliance and Responsible Conduct
Practical Example: Implementing Compliance in a CRISPR Clinical Trial
A team designing a CRISPR trial for a genetic blood disorder took the following steps:
- Submitted a detailed protocol to the IRB, including risk assessments and patient recruitment plans.
- Developed a patient-friendly consent form explaining genome editing and potential risks.
- Established secure databases with limited access to protect patient data.
- Set up a system for reporting adverse events promptly to regulatory bodies.
- Disclosed all financial interests related to the therapy development.
- Conducted regular team training sessions on compliance and ethical conduct.
This structured approach minimized regulatory delays and built confidence among participants and oversight committees.
In summary, responsible genome editing research requires attention to regulatory frameworks, clear communication with participants, rigorous data protection, transparent reporting, conflict of interest disclosure, and ongoing education. These practices not only safeguard participants but also strengthen the quality and credibility of the research.
10. Industrial and Clinical Applications of Genome Editing
10.1 Genome Editing in Agriculture and Food Biotechnology
Genome editing has become a significant tool in agriculture and food biotechnology, allowing precise modifications to crop and livestock genomes. This precision contrasts with traditional breeding, which relies on crossing and selection over multiple generations. Genome editing offers the ability to introduce or remove traits rapidly and with fewer unintended changes.
Applications in Agriculture
Genome editing in agriculture primarily targets traits such as disease resistance, yield improvement, stress tolerance, and nutritional enhancement. For example, editing genes responsible for susceptibility to certain pathogens can create crops that resist diseases without introducing foreign DNA.
One well-known case is the development of mildew-resistant wheat. By knocking out a gene that the fungus exploits to infect the plant, researchers created wheat varieties less vulnerable to powdery mildew. This approach reduces the need for chemical fungicides.
Another example is the modification of tomatoes to extend shelf life. Editing genes involved in the ripening process delays over-ripening and spoilage, which can reduce food waste.
Applications in Food Biotechnology
Beyond crops, genome editing is used in livestock to improve traits like disease resistance, growth rate, and meat quality. For instance, pigs have been edited to resist porcine reproductive and respiratory syndrome (PRRS), a costly viral disease.
In aquaculture, genome editing helps develop fish varieties that grow faster or tolerate environmental stress better, supporting sustainable food production.
Mind Map: Genome Editing Targets in Agriculture and Food Biotechnology
Best Practices in Agricultural Genome Editing
Target Selection: Choose genes with well-characterized functions to minimize unintended effects. For example, targeting susceptibility genes rather than resistance genes can be more effective and stable.
Off-Target Assessment: Use computational tools and experimental validation to confirm edits are specific. In crops like rice, off-target mutations can affect yield or quality, so thorough screening is essential.
Regulatory Compliance: Different countries have varying regulations on genome-edited organisms. Understanding and adhering to these rules is critical for field trials and commercialization.
Trait Validation: Edited plants or animals should undergo phenotypic and molecular testing across multiple generations to ensure trait stability and absence of negative effects.
Example: Developing Disease-Resistant Rice
Researchers targeted the SWEET gene family in rice, which certain bacteria exploit to cause bacterial blight. Using CRISPR-Cas9, they edited promoter regions to prevent bacterial activation without disrupting the gene’s normal function. The edited rice lines showed strong resistance and maintained yield.
Mind Map: Workflow for Developing Genome-Edited Crops
Delivery Methods in Plants
Common delivery methods include Agrobacterium-mediated transformation and biolistics (gene gun). Agrobacterium is preferred for many dicots due to efficiency and lower copy number insertions, while biolistics is often used in monocots like maize.
Example: Enhancing Nutritional Content in Cassava
Cassava is a staple crop with low protein content. By editing genes involved in starch biosynthesis, scientists increased the protein content, improving its nutritional value. This was achieved using CRISPR delivered via Agrobacterium.
Challenges and Considerations
- Regeneration Efficiency: Some crops are difficult to regenerate after transformation, limiting editing success.
- Pleiotropy: Editing one gene may affect multiple traits; careful phenotypic analysis is necessary.
- Public Acceptance: Transparent communication about genome editing methods and safety is important to build trust.
In summary, genome editing in agriculture and food biotechnology provides targeted, efficient ways to improve crops and livestock. Through careful design, validation, and adherence to best practices, it can contribute to sustainable and resilient food systems.
10.2 Clinical Trials Utilizing CRISPR-Based Therapeutics
Clinical trials using CRISPR-based therapeutics represent a critical step in translating genome editing from the lab bench to patient care. These trials test safety, efficacy, and delivery methods in controlled settings, often focusing on monogenic diseases or cancers where gene editing can directly address the underlying cause.
Overview of Clinical Trial Phases
Clinical trials generally progress through phases I to III, each with specific goals:
- Phase I: Primarily assesses safety and dosage in a small group of participants.
- Phase II: Explores efficacy and side effects in a larger cohort.
- Phase III: Confirms effectiveness, monitors adverse reactions, and compares with standard treatments.
CRISPR trials follow this structure but add complexity due to the novelty of genome editing.
Mind Map: Clinical Trial Structure for CRISPR Therapeutics
Types of CRISPR Therapeutics in Trials
-
Ex Vivo Editing: Cells are extracted, edited outside the body, then reintroduced. This approach allows for quality control before infusion.
-
In Vivo Editing: CRISPR components are delivered directly into the patient to edit cells inside the body. This method faces challenges in targeting and delivery.
Example: Ex Vivo Trial for Sickle Cell Disease
One of the earliest CRISPR clinical trials targets sickle cell disease by editing hematopoietic stem cells ex vivo. The goal is to reactivate fetal hemoglobin production by disrupting a regulatory gene, reducing sickling of red blood cells.
- Process: Stem cells are harvested from the patient, edited using CRISPR-Cas9, then infused back.
- Best Practice: Rigorous screening of edited cells ensures minimal off-target mutations before reinfusion.
- Outcome Measures: Hemoglobin levels, frequency of sickle crises, and long-term safety.
Mind Map: Ex Vivo Editing Workflow
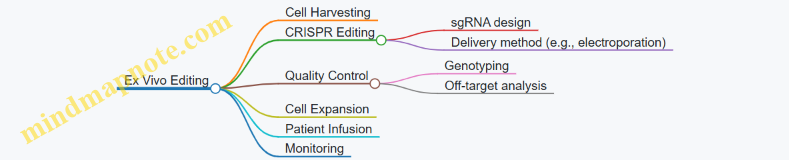
Example: In Vivo Trial for Leber Congenital Amaurosis (LCA)
An in vivo CRISPR trial targets LCA, a genetic eye disorder causing blindness. The CRISPR system is delivered via an adeno-associated virus (AAV) directly into the retina.
- Delivery Challenge: Ensuring the virus reaches target cells without triggering immune responses.
- Safety Measures: Dose escalation studies to identify safe viral loads.
- Efficacy Assessment: Visual acuity tests and retinal imaging.
Mind Map: In Vivo Editing Considerations
Regulatory and Ethical Aspects in CRISPR Trials
Clinical trials must comply with regulatory standards that emphasize patient safety and informed consent. Given the permanent nature of genome edits, long-term follow-up is often mandated.
- Informed Consent: Patients must understand potential risks, including unknown off-target effects.
- Data Transparency: Reporting adverse events promptly is critical.
Example: CAR-T Cell Therapy Trials Using CRISPR
Some trials combine CRISPR editing with CAR-T cell therapies to improve cancer treatment. Editing may knock out genes that cause immune rejection or enhance T cell persistence.
- Editing Targets: PD-1 gene knockout to prevent T cell exhaustion.
- Manufacturing Challenges: Maintaining cell viability and functionality post-editing.
- Outcome Metrics: Tumor response rates and survival.
Mind Map: CRISPR in CAR-T Clinical Trials
Common Challenges in CRISPR Clinical Trials
- Delivery Efficiency: Achieving sufficient editing in target cells.
- Off-Target Effects: Detecting and minimizing unintended edits.
- Immune Responses: Both to delivery vectors and edited cells.
- Scalability: Producing clinical-grade edited cells consistently.
Summary
Clinical trials with CRISPR-based therapeutics are complex undertakings that require careful design, rigorous quality control, and thorough monitoring. Examples from sickle cell disease, inherited blindness, and cancer immunotherapy illustrate different approaches and challenges. Mind maps help visualize workflows and considerations, supporting clearer understanding of these multifaceted studies.
10.3 Manufacturing and Scale-Up of Engineered Cell Therapies
Manufacturing and scale-up of engineered cell therapies is a critical step in translating laboratory successes into treatments accessible to patients. This process involves producing large quantities of cells that are genetically modified, functionally consistent, and safe for clinical use. The challenge lies in maintaining quality and reproducibility while increasing volume.
Key Components of Manufacturing and Scale-Up
Process Development
The process begins with selecting the appropriate cell source, such as autologous T cells or allogeneic stem cells. The choice affects scalability and complexity. For example, autologous therapies require individualized processing, which limits batch size but reduces immune rejection risk.
Vector production, often viral, must be optimized for high titer and purity. Lentiviral vectors are common but require careful handling to ensure safety and consistency.
Culture conditions, including media composition, oxygen levels, and growth factors, directly influence cell expansion and phenotype. For instance, T cell cultures may use IL-2 supplementation to promote proliferation.
Scale-Up Strategies
Scaling up can follow batch or continuous processing. Batch processes are simpler but may face limitations in volume and consistency. Continuous systems, like perfusion bioreactors, allow steady-state production but require more complex control.
Bioreactors come in various forms: stirred-tank, wave, and fixed-bed. Stirred-tank bioreactors are versatile and widely used for suspension cultures, while wave bioreactors offer gentle mixing suited for sensitive cells.
Automation reduces manual variability and contamination risk. Automated systems can handle cell washing, media exchange, and sampling, improving reproducibility.
Quality Control
Quality control ensures that the final cell product meets identity, purity, potency, and safety criteria. Identity testing confirms the cell type and genetic modifications, often using flow cytometry or PCR.
Potency assays measure the functional activity of the cells, such as cytotoxicity for CAR-T cells. Sterility testing detects microbial contamination.
Batch-to-batch consistency is critical. For example, measuring transduction efficiency and cell viability across batches helps maintain standards.
Regulatory Compliance
Manufacturing must comply with Good Manufacturing Practice (GMP) regulations. This includes validated protocols, trained personnel, and thorough documentation.
Validation covers equipment qualification, process validation, and cleaning procedures. Documentation ensures traceability from raw materials to final product.
Logistics
Cryopreservation allows storage and transport of engineered cells. Optimizing freezing protocols preserves viability and function. For example, controlled-rate freezing reduces ice crystal formation.
Supply chain management coordinates raw material sourcing, manufacturing schedules, and distribution to clinical sites.
Example: Manufacturing CAR-T Cells at Scale
- Cell Collection: Leukapheresis collects T cells from the patient.
- Activation and Transduction: T cells are activated with beads and transduced with a lentiviral vector encoding the CAR.
- Expansion: Cells are expanded in a stirred-tank bioreactor with IL-2 supplementation.
- Harvest and Formulation: Cells are harvested, washed, and formulated in infusion media.
- Quality Testing: Identity, potency, sterility, and viability are assessed.
- Cryopreservation: Cells are frozen using controlled-rate freezing.
- Distribution: Frozen cells are shipped to the clinic for infusion.
Each step includes in-process controls to monitor cell growth, transduction efficiency, and phenotype.
Summary
Manufacturing and scale-up of engineered cell therapies require careful coordination of biological, engineering, and regulatory factors. Choosing the right cell source, optimizing culture and vector production, selecting appropriate bioreactors, implementing quality control, and ensuring regulatory compliance are all essential. Practical examples, like CAR-T cell production, illustrate how these elements come together to produce consistent, safe, and effective therapies.
10.4 Quality Control and Standardization in Clinical Applications
Quality control (QC) and standardization are foundational to the clinical application of genome editing and cellular engineering. They ensure that therapies are safe, effective, and reproducible across batches and clinical sites. Without rigorous QC, variability in cell products or gene edits can lead to inconsistent outcomes or safety risks.
Key Components of Quality Control in Genome Editing
- Identity: Confirming that the cell type or engineered product matches the intended specification.
- Purity: Ensuring the absence of unwanted cell types, contaminants, or residual reagents.
- Potency: Demonstrating the functional activity of the edited cells or gene product.
- Safety: Testing for off-target effects, insertional mutagenesis, and absence of pathogens.
- Stability: Verifying that the product maintains its characteristics during storage and handling.
Standardization Challenges
Genome editing introduces complexity because each batch can have unique edits, and cellular products are inherently variable. Standardization involves harmonizing protocols, assays, and acceptance criteria to reduce this variability.
Mind Map: Quality Control Elements in Clinical Genome Editing
Identity Verification
Confirming the identity of the engineered cells is the first step. For example, in CAR-T therapies, flow cytometry is used to verify expression of the CAR construct and T cell markers (CD3, CD4, CD8). Genetic assays such as PCR or sequencing confirm the presence and correct sequence of the intended edit.
Example: A clinical batch of edited hematopoietic stem cells (HSCs) undergoes short tandem repeat (STR) profiling to ensure the cells match the donor and have not been cross-contaminated.
Purity Assessment
Purity testing ensures that the final product is free from unwanted cells or contaminants. This includes testing for residual viral vectors, plasmid DNA, or reagents used during editing.
Example: After CRISPR editing of T cells, residual Cas9 protein and guide RNA are quantified using ELISA and qPCR to confirm clearance before infusion.
Potency Testing
Potency assays measure the functional capacity of the edited cells. These assays must correlate with clinical efficacy.
Example: For engineered T cells, cytotoxicity assays against target tumor cells demonstrate the ability to kill cancer cells. In gene correction therapies, restoration of protein function can be measured by enzymatic activity or phenotypic rescue.
Safety Testing
Safety testing is critical to detect unintended effects. Off-target editing can cause mutations with unknown consequences.
Example: GUIDE-seq or targeted deep sequencing is used to identify off-target sites in edited cells. Additionally, tests for replication-competent viruses and endotoxins are performed.
Stability Testing
Stability studies confirm that the product maintains quality attributes during storage and transport.
Example: Cryopreserved CAR-T cells are thawed at different time points to assess viability, phenotype, and function, ensuring they remain effective after shipment.
Mind Map: Standardization Strategies
Protocol Harmonization
Standardizing editing protocols reduces batch-to-batch variability. This includes consistent guide RNA design, delivery methods, and culture conditions.
Example: A manufacturing facility uses a validated electroporation protocol with fixed parameters for all T cell editing runs.
Assay Validation
All QC assays must be validated to ensure they reliably measure the intended attribute.
Example: A flow cytometry assay for CAR expression is validated for linearity, precision, and limit of detection before use in QC.
Acceptance Criteria
Clear, predefined criteria determine whether a batch passes QC.
Example: Edited cells must express CAR on at least 80% of viable cells, have less than 5% residual Cas9 protein, and demonstrate >50% target cell killing in potency assays.
Documentation and Training
Accurate records and trained personnel ensure traceability and reproducibility.
Example: Batch manufacturing records include detailed steps, reagent lot numbers, and QC results. Staff undergo regular competency assessments.
Summary
Quality control and standardization in clinical genome editing require a multi-pronged approach. Each QC element—identity, purity, potency, safety, and stability—must be rigorously tested with validated assays. Standardization of protocols and clear acceptance criteria help ensure consistent, safe, and effective therapies. Concrete examples from CAR-T and stem cell editing illustrate how these principles apply in practice.
10.5 Example: Manufacturing CAR-T Cells for Clinical Use
Manufacturing CAR-T cells for clinical use is a multi-step process that transforms a patient’s own T cells into targeted cancer fighters. The process requires precision, sterility, and quality control to ensure safety and efficacy. Below is a detailed overview of the key steps, accompanied by mind maps and concrete examples.
Step 1: Collection of Patient T Cells (Leukapheresis)
The process begins with leukapheresis, where peripheral blood mononuclear cells (PBMCs), including T cells, are collected from the patient. This is done using a machine that separates white blood cells from the blood and returns the rest to the patient.
- Best Practice: Ensure patient hydration and monitor cell counts to optimize yield.
Step 2: T Cell Enrichment and Activation
Once collected, T cells are isolated from the leukapheresis product. Magnetic bead-based separation targeting CD3 or CD4/CD8 markers is commonly used.
Activation is critical to prepare T cells for genetic modification. This usually involves anti-CD3/CD28 antibody-coated beads that mimic antigen-presenting cells, stimulating T cell proliferation.
- Example: Using Dynabeads® CD3/CD28 for activation results in consistent T cell expansion.
Step 3: Genetic Modification (Transduction or Transfection)
The activated T cells are genetically engineered to express the chimeric antigen receptor (CAR). Viral vectors, typically lentivirus or retrovirus, are the most common delivery systems.
-
Best Practice: Use a vector with a strong promoter and safety features like self-inactivating long terminal repeats (SIN-LTRs).
-
Example: Lentiviral transduction at a multiplicity of infection (MOI) of 5 can achieve >70% CAR expression with minimal toxicity.
Step 4: Expansion of CAR-T Cells
After transduction, cells are cultured in media supplemented with cytokines such as IL-2, IL-7, or IL-15 to promote proliferation.
-
Best Practice: Monitor cell density and viability daily; split cultures to prevent overconfluence.
-
Example: A 10-day expansion protocol can yield 1-5 billion CAR-T cells, sufficient for therapeutic dosing.
Step 5: Harvesting and Formulation
Expanded CAR-T cells are harvested, washed to remove residual beads and cytokines, and formulated in a cryopreservation solution.
- Best Practice: Use controlled-rate freezing to maintain cell viability during storage.
Step 6: Quality Control and Release Testing
Before infusion, CAR-T products undergo rigorous testing:
-
Identity: Confirm CAR expression by flow cytometry.
-
Purity: Assess contamination by other cell types or microbial agents.
-
Potency: Functional assays like cytokine release or cytotoxicity against target cells.
-
Safety: Test for replication-competent virus and endotoxin levels.
-
Example: A release criterion might require >50% CAR-positive T cells and <5% dead cells.
Mind Map: CAR-T Cell Manufacturing Workflow
Mind Map: Quality Control Parameters
Example Scenario: Manufacturing CAR-T Cells for a Patient with B-cell Lymphoma
- Leukapheresis is performed, yielding 2 x 10^9 PBMCs.
- T cells are enriched using CD3 magnetic beads, resulting in 5 x 10^8 T cells.
- Cells are activated with anti-CD3/CD28 beads and transduced with a lentiviral vector encoding an anti-CD19 CAR at MOI 5.
- After 10 days of expansion with IL-2, the culture reaches 2 x 10^9 CAR-positive T cells.
- Cells are harvested, washed, and cryopreserved.
- Quality control confirms 65% CAR expression, >90% viability, no microbial contamination, and functional cytotoxicity against CD19+ target cells.
- The product is released for infusion.
This example highlights the importance of each step’s optimization and monitoring to produce a safe and effective CAR-T cell therapy.
In summary, manufacturing CAR-T cells involves coordinated steps from patient cell collection to final product release. Each phase requires specific best practices and quality checks to ensure the therapy meets clinical standards. The process combines cell biology, genetic engineering, and manufacturing principles to deliver personalized medicine.
11. Troubleshooting and Optimization in Genome Editing Experiments
11.1 Common Challenges in Genome Editing Protocols
Genome editing protocols, while powerful, come with a set of recurring challenges that can complicate experiments and affect outcomes. Understanding these issues upfront helps in designing better experiments and troubleshooting problems efficiently.
Target Site Selection and sgRNA Design
Choosing the right target site is critical. Poorly designed single-guide RNAs (sgRNAs) can lead to low editing efficiency or unintended off-target effects.
- Challenge: Identifying target sequences that balance high on-target activity with minimal off-target potential.
- Example: An sgRNA targeting a gene with multiple homologous regions may cause edits in unintended loci, complicating data interpretation.
Mind Map: Target Site Selection
Delivery of Editing Components
Efficient delivery of CRISPR components (Cas protein, sgRNA, donor DNA) into cells is often a bottleneck.
- Challenge: Different cell types vary in their susceptibility to delivery methods.
- Example: Primary T cells are notoriously difficult to transfect, requiring electroporation or viral vectors, each with trade-offs in toxicity and efficiency.
Mind Map: Delivery Challenges
Off-Target Effects
Unintended edits at genomic sites similar to the target sequence can cause unwanted mutations.
- Challenge: Detecting and minimizing off-target activity without compromising on-target efficiency.
- Example: Editing in neuronal cells where off-target mutations could alter cell function or viability.
Mind Map: Off-Target Effects
Cellular Response and Toxicity
Genome editing can trigger DNA damage responses, apoptosis, or immune reactions.
- Challenge: Balancing editing efficiency with cell survival.
- Example: High Cas9 expression levels may increase editing but also cause cytotoxicity in stem cells.
Mind Map: Cellular Response
Repair Pathway Bias
After DNA cleavage, cells repair breaks via non-homologous end joining (NHEJ) or homology-directed repair (HDR), influencing the type of edit.
- Challenge: HDR is less efficient and limited to certain cell cycle phases.
- Example: Attempting precise gene correction in quiescent cells often results in indels due to predominant NHEJ.
Mind Map: DNA Repair Pathways
Clonal Variation and Mosaicism
Edited cell populations can be heterogeneous, with varying genotypes and phenotypes.
- Challenge: Isolating and characterizing clones with desired edits.
- Example: In edited stem cell cultures, some clones may carry biallelic knockouts while others remain unedited.
Mind Map: Clonal Variation
Experimental Controls and Validation
Proper controls are essential to distinguish editing effects from background noise.
- Challenge: Designing negative and positive controls that accurately reflect experimental conditions.
- Example: Using a non-targeting sgRNA control to assess off-target toxicity.
Mind Map: Controls and Validation
Summary Example: Editing a Disease Gene in Fibroblasts
Suppose you want to knock out a gene implicated in fibrosis using CRISPR-Cas9 in primary human fibroblasts. Challenges include designing sgRNAs that avoid off-targets in gene families, delivering components efficiently without killing cells, and confirming edits in a heterogeneous population. Using electroporation with optimized pulse settings, high-fidelity Cas9, and single-cell cloning can help overcome these hurdles.
Understanding these common challenges and planning accordingly improves the chances of successful genome editing experiments.
11.2 Optimizing Editing Efficiency Across Cell Types
Optimizing genome editing efficiency across different cell types requires a clear understanding of the unique biological and physical characteristics of each cell population. Editing efficiency depends on factors such as cell cycle stage, chromatin accessibility, DNA repair pathways, and the method of delivering editing components. This section breaks down these factors and provides practical examples and mind maps to clarify the optimization process.
Key Factors Influencing Editing Efficiency
- Cell Type Characteristics: Primary cells, stem cells, immortalized cell lines, and differentiated cells each respond differently to editing tools.
- Cell Cycle Stage: Some editing outcomes depend on whether cells are in G1, S, or G2/M phases.
- Chromatin State: Open chromatin is generally more accessible to editing machinery.
- DNA Repair Pathways: The balance between non-homologous end joining (NHEJ) and homology-directed repair (HDR) affects editing outcomes.
- Delivery Method: Electroporation, viral vectors, lipid nanoparticles, and microinjection vary in efficiency and toxicity depending on cell type.
Mind Map: Factors Affecting Editing Efficiency
Practical Considerations and Examples
Cell Type-Specific Optimization
Primary cells often have lower editing efficiencies than immortalized lines due to limited proliferation and sensitivity to manipulation. For example, editing T cells requires careful activation and culture conditions to maintain viability and function. In contrast, HEK293 cells tolerate electroporation well and often show high editing rates.
Example: When editing human hematopoietic stem cells (HSCs), researchers typically pre-stimulate cells with cytokines to encourage cycling, which improves HDR rates. Without this step, HDR-mediated edits drop significantly.
Cell Cycle Synchronization
HDR is most active during the S and G2 phases, while NHEJ operates throughout the cell cycle. Synchronizing cells or selecting cells in S/G2 can increase precise editing.
Example: In induced pluripotent stem cells (iPSCs), synchronization with thymidine block before CRISPR delivery enhanced HDR efficiency by 2-3 fold compared to asynchronous populations.
Chromatin Accessibility
Target sites in open chromatin regions are more accessible to Cas nucleases. Chromatin modifiers or small molecules can transiently open chromatin to improve editing.
Example: Treatment of fibroblasts with histone deacetylase inhibitors prior to editing increased Cas9 access and editing efficiency at a previously refractory locus.
Delivery Method Optimization
Choosing the right delivery method is critical. Electroporation is effective for many cell types but can cause toxicity in sensitive cells. Viral vectors offer high efficiency but have size limitations and safety concerns.
Example: Lipid nanoparticle delivery of Cas9 mRNA and guide RNA achieved efficient editing in primary hepatocytes with lower toxicity than electroporation.
Mind Map: Delivery Method Considerations
Summary of Optimization Steps
- Characterize the cell type: Understand growth rate, sensitivity, and DNA repair preferences.
- Choose delivery method: Balance efficiency and toxicity for the specific cell.
- Consider cell cycle: Synchronize or enrich for phases favoring desired repair pathways.
- Modify chromatin if needed: Use small molecules to enhance accessibility.
- Validate and iterate: Test editing efficiency and cell viability, then adjust parameters.
Example Workflow: Editing Human T Cells
- Activate T cells with CD3/CD28 beads to induce proliferation.
- Deliver Cas9 RNP complexes via electroporation using optimized pulse settings.
- Culture cells in IL-2 containing media to support growth.
- Assess editing efficiency by flow cytometry and sequencing.
This approach typically yields editing efficiencies above 70% with high viability.
Optimizing editing efficiency is a multi-factorial process that requires tailoring protocols to the biology of each cell type. Systematic testing and adjustment of delivery methods, cell cycle conditions, and chromatin state can substantially improve outcomes.
11.3 Addressing Delivery Failures and Cytotoxicity
Delivery failures and cytotoxicity are two common hurdles in genome editing experiments. Efficient delivery of editing components into target cells is essential, but the methods used can sometimes harm cells or fail to introduce the material effectively. Understanding the causes and solutions for these issues can improve experimental outcomes.
Causes of Delivery Failures
- Incompatible delivery method: Different cell types respond differently to delivery methods. For example, electroporation works well for some suspension cells but can be lethal for sensitive primary cells.
- Poor quality or degraded reagents: Plasmids, RNA, or protein components that are degraded or contaminated reduce delivery efficiency.
- Suboptimal dosing: Too little editing material results in low uptake; too much can cause toxicity.
- Inefficient targeting: Delivery vehicles may not reach the intended cells due to poor tropism or uptake mechanisms.
- Physical barriers: Cell membrane composition, extracellular matrix, or cell density can impede delivery.
Causes of Cytotoxicity
- Physical damage: Electroporation and microinjection can physically disrupt membranes.
- Chemical toxicity: Lipid nanoparticles or chemical transfection reagents may be toxic at high concentrations.
- Immune activation: Viral vectors or foreign nucleic acids can trigger immune responses.
- Off-target effects: Unintended genome edits can induce cell stress or death.
- Overexpression toxicity: Excessive expression of Cas proteins or guide RNAs can overwhelm cellular machinery.
Mind Map: Delivery Failures and Cytotoxicity
Strategies to Address Delivery Failures
-
Match delivery method to cell type:
- Use electroporation for robust cell lines like HEK293.
- Use lipid nanoparticles or viral vectors for primary or stem cells.
-
Optimize reagent quality:
- Use freshly prepared, endotoxin-free plasmids.
- Confirm RNA integrity with gel electrophoresis.
-
Titrate doses carefully:
- Start with low doses and increase gradually.
- Monitor cell viability alongside editing efficiency.
-
Enhance targeting efficiency:
- Use cell-specific promoters or targeting ligands on delivery vehicles.
- Modify vector pseudotypes to improve tropism.
-
Modify culture conditions:
- Reduce cell density to improve access.
- Use enzymatic treatments to loosen extracellular matrix.
Strategies to Mitigate Cytotoxicity
-
Adjust physical delivery parameters:
- Lower voltage or pulse length in electroporation.
- Use gentler microinjection techniques.
-
Select less toxic reagents:
- Compare different lipid formulations.
- Use recombinant Cas9 protein instead of plasmid to reduce prolonged expression.
-
Include immunosuppressive agents if appropriate:
- Use transient immunosuppressants in sensitive primary cells.
-
Limit expression duration:
- Use transient delivery methods (mRNA, RNP complexes).
- Employ inducible promoters.
-
Monitor and minimize off-target activity:
- Use high-fidelity Cas9 variants.
- Design highly specific guide RNAs.
Mind Map: Solutions to Delivery Failures and Cytotoxicity
Examples
Example 1: Electroporation in Primary T Cells
Primary human T cells are notoriously sensitive to electroporation. Initial attempts using standard parameters resulted in low viability (~40%) and poor editing efficiency. By reducing the voltage and pulse duration, viability improved to 75%, with a modest increase in editing efficiency. Additionally, switching from plasmid DNA to Cas9 RNP complexes reduced cytotoxicity further and shortened editing time.
Example 2: Lipid Nanoparticle Delivery in Hepatocytes
Lipid nanoparticles (LNPs) are popular for in vivo delivery but can be toxic at high doses. In hepatocyte cultures, high LNP concentrations caused cell rounding and death. By optimizing lipid composition and lowering the dose, cells maintained normal morphology and showed efficient genome editing. Including polyethylene glycol (PEG) lipids reduced aggregation and improved biocompatibility.
Example 3: Viral Vector Tropism Modification
A lentiviral vector designed for broad tropism failed to transduce neural progenitor cells efficiently. By pseudotyping the vector with a rabies virus glycoprotein, transduction efficiency increased threefold without added toxicity. This targeted approach improved delivery success while preserving cell health.
Summary
Addressing delivery failures and cytotoxicity requires a combination of understanding the biology of the target cells, the properties of the delivery method, and the nature of the editing components. Systematic optimization, including dose titration, reagent quality control, and method tailoring, can significantly improve outcomes. Monitoring cell health alongside editing efficiency is essential to balance effectiveness with viability.
11.4 Data Analysis Pitfalls and Solutions
Genome editing experiments generate complex datasets that require careful analysis to draw valid conclusions. Missteps in data handling or interpretation can lead to misleading results or missed insights. This section highlights common pitfalls encountered during data analysis and offers practical solutions, illustrated with clear examples and mind maps.
Common Data Analysis Pitfalls
- Ignoring Experimental Context: Treating raw data without considering experimental design or biological variability.
- Overlooking Controls: Failing to include or properly analyze negative and positive controls.
- Misinterpreting Off-Target Effects: Confusing background noise or sequencing errors with true off-target edits.
- Inadequate Statistical Testing: Using inappropriate tests or neglecting multiple testing corrections.
- Data Normalization Errors: Applying incorrect normalization methods that distort true signal.
- Selective Reporting: Cherry-picking data points that support hypotheses while ignoring contradictory evidence.
Mind Map: Data Analysis Pitfalls
Pitfall 1: Ignoring Experimental Context
Data without context is like a map without landmarks. For example, analyzing CRISPR editing efficiency without considering cell type or passage number can skew results. Different cell lines may respond differently to the same editing protocol, affecting indel rates or expression changes.
Solution: Always integrate metadata such as cell type, passage, and treatment conditions into your analysis pipeline. Use replicates to capture biological variability and avoid overinterpreting single-sample results.
Pitfall 2: Overlooking Controls
Controls anchor your analysis. Imagine measuring editing efficiency without a non-edited control; you risk mistaking sequencing errors for edits.
Example: In a CRISPR experiment targeting a gene, sequencing the unedited control sample helps establish baseline noise. Without it, low-frequency variants might be misclassified as off-target edits.
Solution: Include and analyze proper controls. Compare edited samples against unedited or mock-treated controls to distinguish true edits from artifacts.
Pitfall 3: Misinterpreting Off-Target Effects
Off-target analysis can be tricky. Sequencing errors or PCR artifacts sometimes mimic off-target mutations.
Example: A low-frequency variant appearing in only one replicate might be a sequencing error rather than a genuine off-target event.
Solution: Use replicates and orthogonal validation methods. Confirm off-target sites with independent assays like targeted deep sequencing or GUIDE-seq. Apply stringent filters to exclude variants below error thresholds.
Mind Map: Off-Target Data Interpretation
Pitfall 4: Inadequate Statistical Testing
Using the wrong statistical test or ignoring multiple comparisons can inflate false positives.
Example: Testing editing efficiency across multiple loci without adjusting p-values can make insignificant differences appear significant.
Solution: Choose tests appropriate for your data distribution (e.g., t-test, Mann-Whitney). Apply corrections like Bonferroni or Benjamini-Hochberg when testing multiple hypotheses.
Pitfall 5: Data Normalization Errors
Normalization adjusts for technical variation but can introduce bias if misapplied.
Example: Normalizing gene expression data by total read count without accounting for highly expressed genes can mask true expression changes.
Solution: Select normalization methods suited to your data type. For RNA-seq, consider methods like TPM or DESeq2’s median ratio normalization. Validate normalization by checking that control genes remain stable.
Pitfall 6: Selective Reporting
Cherry-picking favorable data undermines scientific integrity.
Example: Reporting only samples with high editing efficiency while ignoring failed replicates gives a skewed impression.
Solution: Present complete datasets, including negative or inconclusive results. Transparency builds trust and helps others reproduce findings.
Summary Mind Map: Solutions to Data Analysis Pitfalls
Data analysis is as important as the experimental work itself. Recognizing these pitfalls and applying the suggested solutions will improve the reliability of your conclusions and the overall quality of your genome editing research.
11.5 Practical Troubleshooting Guide with Real-World Examples
Troubleshooting genome editing experiments can feel like solving a puzzle with many moving parts. The key is to systematically identify where the process is breaking down and apply targeted fixes. Below, we break down common issues, their causes, and practical solutions, supported by examples from real lab scenarios.
Mind Map: Troubleshooting Genome Editing Experiments
Low Editing Efficiency
Problem: After transfection, only a small fraction of cells show the desired edit.
Common Causes and Solutions:
-
Poor sgRNA Design: sgRNAs with low on-target activity or high off-target potential reduce editing. Use design tools focusing on target specificity and efficiency. Example: In a study targeting the HBB gene, redesigning sgRNAs based on updated scoring algorithms increased editing from 10% to 45%.
-
Inefficient Delivery: Delivery method may not suit the cell type. For example, electroporation works well for hematopoietic stem cells but may kill sensitive neurons. Switching to lipid nanoparticles improved editing efficiency in primary neurons from 5% to 30%.
-
Cell Cycle Stage: Homology-directed repair (HDR) is more active in S/G2 phases. Synchronizing cells or using cell cycle modulators can enhance HDR-based edits. Example: Synchronizing HEK293 cells increased HDR efficiency by 20%.
-
DNA Repair Pathway Activity: Some cells favor non-homologous end joining (NHEJ), limiting precise edits. Using small molecule inhibitors of NHEJ (e.g., SCR7) can tip the balance toward HDR.
High Off-Target Effects
Problem: Unintended edits appear at non-target sites.
Common Causes and Solutions:
-
sgRNA Specificity Issues: sgRNAs with partial matches elsewhere in the genome cause off-target cuts. Designing sgRNAs with minimal homology to other loci reduces this risk.
-
Excessive Nuclease Expression: High levels of Cas9 increase off-target activity. Using ribonucleoprotein (RNP) delivery or inducible expression systems limits nuclease exposure.
-
Inadequate Validation: Skipping off-target screening can miss problems. Employing techniques like GUIDE-seq or targeted deep sequencing confirms specificity.
Example: A lab reduced off-target mutations in T cells by switching from plasmid-based Cas9 expression to RNP delivery, cutting off-target edits by over 70%.
Delivery Failures
Problem: No or poor uptake of editing components.
Common Causes and Solutions:
-
Vector Toxicity: Viral vectors can be toxic or immunogenic. Testing different serotypes or reducing viral load can help.
-
Suboptimal Transfection Conditions: Electroporation voltage or reagent ratios might be off. Optimizing parameters for each cell type is essential.
-
Cell Type Resistance: Some cells are inherently hard to transfect. Using viral delivery or cell-penetrating peptides may be necessary.
Example: Primary fibroblasts showed <5% transfection with lipofection but improved to 60% using nucleofection.
Cytotoxicity
Problem: Cells die after editing attempts.
Common Causes and Solutions:
-
Overexpression of Editing Components: High Cas9 or sgRNA levels can stress cells. Titrating component amounts reduces toxicity.
-
Delivery Reagent Toxicity: Some reagents harm cells. Testing multiple reagents and including controls helps identify the culprit.
-
Immune Response Activation: In primary immune cells, foreign proteins can trigger responses. Using humanized Cas9 variants or transient delivery reduces this.
Example: Reducing Cas9 plasmid concentration by half improved viability in primary T cells from 40% to 75%.
Validation Problems
Problem: Difficulty confirming edits.
Common Causes and Solutions:
-
PCR Amplification Failure: Large deletions or complex edits can hinder PCR. Designing primers flanking smaller regions or using long-range PCR can help.
-
Insufficient Sequencing Depth: Low coverage misses rare edits. Increasing sequencing depth or using targeted amplicon sequencing improves detection.
-
Misinterpretation of Results: Mixed cell populations complicate analysis. Single-cell sequencing or clonal isolation clarifies outcomes.
Example: In editing iPSCs, subcloning and Sanger sequencing of individual clones revealed precise edits masked in bulk sequencing.
Summary Table of Troubleshooting Steps
| Issue | Cause | Action to Take | Example Outcome |
|---|---|---|---|
| Low Efficiency | Poor sgRNA design | Redesign sgRNA with improved specificity scores | Editing improved from 10% to 45% |
| Inefficient delivery | Switch delivery method (e.g., lipid nanoparticles) | Primary neurons editing increased to 30% | |
| High Off-Target | Excessive nuclease expression | Use RNP delivery or inducible systems | Off-target reduced by 70% |
| Delivery Failure | Suboptimal transfection | Optimize electroporation parameters | Fibroblast transfection improved to 60% |
| Cytotoxicity | Overexpression of components | Reduce plasmid or reagent concentration | T cell viability improved from 40% to 75% |
| Validation Issues | PCR failure | Design alternative primers or use long-range PCR | Successful amplification and confirmation |
By approaching troubleshooting with a clear framework and concrete examples, you can systematically identify bottlenecks and improve your genome editing experiments. Remember, no single fix works for all cases; iterative testing and careful documentation are your best allies.
12. Case Studies in Genome Editing and Therapeutic Design
12.1 CRISPR Correction of Cystic Fibrosis Mutations
Cystic fibrosis (CF) is a genetic disorder caused primarily by mutations in the CFTR gene, which encodes a chloride channel critical for maintaining fluid balance in epithelial tissues. The most common mutation, ΔF508, results in a misfolded protein that is degraded before reaching the cell surface. CRISPR-based genome editing offers a targeted approach to correct such mutations at the DNA level, potentially restoring normal CFTR function.
Understanding the Mutation Landscape
Before attempting correction, it is essential to characterize the specific mutation(s) present. CFTR mutations fall into several classes, including those causing defective protein production, processing, gating, or conductance. Each class may require a different editing strategy.
Mind Map: CFTR Mutation Classes and Editing Strategies
Designing the CRISPR Correction
The first step is to design guide RNAs (gRNAs) that target sequences near the mutation site. For ΔF508, this involves targeting exon 11 of CFTR. The choice of gRNA must balance efficiency and specificity to minimize off-target effects.
Next, a repair template is introduced to guide homology-directed repair (HDR). This template contains the correct sequence to replace the mutation. In practice, single-stranded oligodeoxynucleotides (ssODNs) are often used for small edits.
Mind Map: CRISPR Correction Workflow for CFTR ΔF508
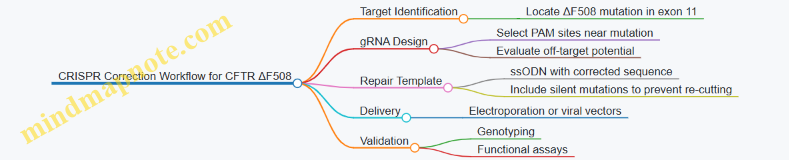
Delivery and Editing in Relevant Cell Types
Primary airway epithelial cells or induced pluripotent stem cells (iPSCs) derived from CF patients are common models. Delivery methods include electroporation of ribonucleoprotein complexes (Cas9 protein + gRNA) along with the repair template.
For example, in a study correcting ΔF508 in patient-derived iPSCs, electroporation of Cas9 RNPs and ssODNs achieved editing efficiencies around 20-30%. Edited cells were then differentiated into airway epithelial cells to assess CFTR function.
Validation of Correction
Validation involves confirming the DNA sequence correction and assessing functional restoration.
- Genotyping: PCR amplification of the target region followed by sequencing confirms the presence of the corrected allele.
- Functional Assays: Chloride ion transport assays, such as Ussing chamber measurements, evaluate restored CFTR channel activity.
Mind Map: Validation Steps
Example: Correcting ΔF508 in Patient-Derived iPSCs
- Cell Preparation: iPSCs from a CF patient harboring ΔF508 are cultured.
- CRISPR Components: Cas9 protein and gRNA targeting exon 11 are complexed.
- Repair Template: ssODN with the wild-type sequence and silent mutations to prevent Cas9 re-cutting is synthesized.
- Delivery: Electroporation introduces the RNP and ssODN into iPSCs.
- Selection and Expansion: Single-cell clones are expanded and screened.
- Validation: Sequencing confirms correction; differentiated epithelial cells show restored chloride transport.
This example illustrates how precise editing can address a common CF mutation.
Best Practices Summary
- gRNA Design: Use computational tools to minimize off-target activity and select guides close to the mutation.
- Repair Template: Incorporate silent mutations to avoid repeated cutting and improve HDR efficiency.
- Delivery Method: Optimize for cell type; RNP electroporation reduces persistent nuclease expression.
- Validation: Combine molecular and functional assays to confirm correction.
- Off-Target Monitoring: Conduct targeted sequencing to ensure genome integrity.
By integrating these practices, researchers can effectively apply CRISPR to correct CFTR mutations, advancing therapeutic development for cystic fibrosis.
12.2 Engineering Beta Cells for Diabetes Treatment
Diabetes, particularly type 1 diabetes, results from the loss or dysfunction of insulin-producing beta cells in the pancreas. Engineering beta cells aims to restore insulin production and regulation, offering a potential therapeutic avenue. This section focuses on the methods, challenges, and examples of beta cell engineering using genome editing and cellular engineering techniques.
Understanding Beta Cell Function and Dysfunction
Beta cells sense blood glucose levels and secrete insulin accordingly. Their dysfunction or destruction disrupts glucose homeostasis, leading to diabetes. Engineering efforts focus on either repairing existing beta cells, generating new functional beta cells from stem cells, or protecting transplanted cells from immune attack.
Key Strategies in Beta Cell Engineering
- Gene Correction: Fixing mutations in beta cells that impair function.
- Cell Replacement: Differentiating stem cells into beta-like cells.
- Immune Evasion: Modifying cells to avoid immune system destruction.
Mind Map: Beta Cell Engineering Components
Gene Correction in Beta Cells
Using CRISPR-Cas9, specific mutations causing monogenic diabetes can be corrected in patient-derived induced pluripotent stem cells (iPSCs). For example, mutations in the INS gene or transcription factors like PDX1 can be repaired. After correction, these iPSCs are differentiated into beta-like cells, restoring insulin secretion capacity.
Example: A patient with a mutation in the INS gene had their iPSCs edited using CRISPR to correct the mutation. Post-editing, the cells differentiated into beta-like cells that secreted insulin in response to glucose, demonstrating functional rescue.
Differentiation of Stem Cells into Beta-like Cells
Protocols exist to guide pluripotent stem cells through developmental stages mimicking pancreatic development. This involves sequential exposure to growth factors and signaling molecules to induce definitive endoderm, pancreatic progenitors, and finally beta-like cells.
Best Practice: Monitor marker expression at each stage (e.g., SOX17 for endoderm, PDX1 for pancreatic progenitors, and INS for beta cells) to ensure proper differentiation.
Example: Human embryonic stem cells were differentiated into beta-like cells over 20 days using a stepwise protocol. The resulting cells secreted insulin in response to glucose challenges in vitro.
Engineering Immune Evasion
One major hurdle is immune rejection of transplanted beta cells. Genome editing can knock out HLA class I and II genes to reduce immune recognition. Alternatively, cells can be engineered to express immune checkpoint proteins like PD-L1 to suppress immune attack.
Example: Beta-like cells derived from edited iPSCs lacking HLA-A and HLA-B showed reduced recognition by cytotoxic T cells in co-culture assays.
Delivery and Transplantation
Encapsulation devices can protect engineered beta cells from immune cells while allowing nutrient and insulin exchange. Alternatively, direct transplantation into the liver or subcutaneous space is performed.
Example: Encapsulated beta-like cells implanted in diabetic mice restored normoglycemia for several weeks without immunosuppression.
Functional Validation of Engineered Beta Cells
Testing insulin secretion in response to glucose is essential. Static incubation assays and perifusion systems measure insulin release dynamics. Additionally, transplantation into diabetic animal models assesses in vivo functionality.
Example: Engineered beta cells transplanted into diabetic mice normalized blood glucose levels within two weeks, confirmed by glucose tolerance tests.
Mind Map: Workflow for Engineering Beta Cells
Summary
Engineering beta cells for diabetes treatment combines genome editing, stem cell biology, and immunology. Each step requires careful design and validation to ensure functional insulin secretion and immune compatibility. Concrete examples demonstrate how CRISPR-mediated gene correction and stem cell differentiation can restore beta cell function, while immune engineering addresses rejection challenges.
12.3 Genome Editing in Hematopoietic Stem Cells (HSCs)
Hematopoietic stem cells (HSCs) are the blood-forming stem cells residing primarily in the bone marrow. They have the unique ability to self-renew and differentiate into all blood cell types. This makes them a prime target for genome editing aimed at treating blood disorders, immunodeficiencies, and certain cancers.
Why Edit HSCs?
Editing HSCs allows permanent correction of genetic defects in the blood system. Since HSCs repopulate the entire hematopoietic system, a successful edit can lead to long-term therapeutic effects. For example, editing the gene responsible for sickle cell disease in HSCs can potentially cure the patient by producing healthy red blood cells.
Challenges in Editing HSCs
- Maintaining Stemness: HSCs are sensitive and can lose their stem-like properties during culture and manipulation.
- Efficient Delivery: Introducing genome editing components without harming the cells is tricky.
- Off-Target Effects: Unintended edits can have serious consequences.
- Engraftment: Edited cells must successfully engraft and repopulate the bone marrow.
Workflow Overview
Step 1: Isolation and Culture
HSCs are typically isolated from bone marrow, peripheral blood after mobilization, or umbilical cord blood. Purity is critical; markers like CD34+ and CD38- help identify true HSCs. Culture conditions aim to keep cells quiescent or minimally proliferative to preserve stemness while allowing enough activity for editing.
Step 2: Choosing the Editing Tool
CRISPR-Cas9 is the most common tool due to its simplicity and efficiency. Alternatives like base editors or prime editors offer more precise changes without double-strand breaks, which can be advantageous in sensitive HSCs.
Step 3: Delivery Methods
- Electroporation of Ribonucleoprotein (RNP): Direct delivery of Cas9 protein complexed with guide RNA reduces exposure time and off-target effects.
- Viral Vectors: Lentivirus or AAV can deliver editing components or donor templates but carry risks of insertional mutagenesis.
- Lipid Nanoparticles: Emerging as a non-viral option but less common in HSCs.
Example: Electroporation of Cas9 RNP with a single-stranded DNA donor template has been shown to achieve efficient homology-directed repair in HSCs with minimal toxicity.
Step 4: Validation and Expansion
After editing, cells undergo genotyping to confirm the intended edit and check for off-target mutations. Functional assays test the ability of edited HSCs to differentiate into multiple lineages. Expansion protocols aim to increase cell numbers without losing stemness.
Step 5: Transplantation and Monitoring
Edited HSCs are transplanted back into the patient or animal models. Monitoring includes tracking engraftment efficiency, lineage reconstitution, and long-term safety.
Example Case: Editing the Beta-Globin Gene
Sickle cell disease is caused by a mutation in the beta-globin gene (HBB). Editing HSCs to correct this mutation or reactivate fetal hemoglobin genes can alleviate symptoms.
- Guide RNA Design: Targeting the sickle mutation site or regulatory regions of fetal hemoglobin.
- Delivery: Electroporation of Cas9 RNP and donor DNA.
- Validation: PCR and sequencing confirm correction; flow cytometry measures hemoglobin expression.
- Outcome: Edited HSCs produce healthy red blood cells upon transplantation.
Best Practices
- Use freshly isolated or minimally cultured HSCs to preserve stemness.
- Optimize electroporation parameters to balance editing efficiency and viability.
- Design highly specific guide RNAs using computational tools and validate off-targets experimentally.
- Include functional assays alongside genotyping to confirm therapeutic potential.
- Monitor long-term engraftment and safety in relevant models.
Mind Map: Best Practices in HSC Genome Editing
By carefully integrating these steps and considerations, genome editing in HSCs can be performed effectively and safely, enabling therapeutic applications that address a range of hematological diseases.
12.4 Cellular Engineering for Neurodegenerative Disease Models
Neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s disease involve progressive loss of neuronal function and structure. Modeling these diseases at the cellular level helps researchers understand disease mechanisms and test potential therapies. Cellular engineering offers tools to create accurate, manipulable models that reflect key aspects of neurodegeneration.
Key Objectives in Cellular Engineering for Neurodegenerative Models
- Reproduce disease-relevant phenotypes in vitro
- Introduce or correct genetic mutations associated with disease
- Generate specific neuronal subtypes affected in the disease
- Enable functional assays to measure neuronal health and activity
Approaches to Cellular Engineering
-
Induced Pluripotent Stem Cells (iPSCs)
- Patient-derived iPSCs retain genetic background, including disease mutations.
- Differentiation protocols guide iPSCs into neurons or glial cells relevant to the disease.
- Genome editing tools like CRISPR can introduce or correct mutations to create isogenic controls.
-
Direct Reprogramming
- Converts somatic cells directly into neurons, bypassing pluripotency.
- Useful for rapid generation of disease-relevant neurons.
-
Genome Editing
- CRISPR-Cas9 and base editors introduce precise mutations.
- Enables study of specific gene variants and their effects on neuronal function.
-
3D Culture Systems and Organoids
- Mimic brain tissue architecture.
- Allow study of cell-cell interactions and complex phenotypes.
Mind Map: Cellular Engineering Strategies for Neurodegenerative Disease Models
Example: Modeling Parkinson’s Disease Using iPSC-Derived Dopaminergic Neurons
Parkinson’s disease primarily affects dopaminergic neurons in the substantia nigra. Researchers obtain skin fibroblasts from a patient carrying a mutation in the LRRK2 gene, reprogram them into iPSCs, then differentiate into dopaminergic neurons. Using CRISPR-Cas9, they generate an isogenic control line by correcting the mutation. Comparing the two lines reveals differences in mitochondrial function and susceptibility to oxidative stress, which are hallmarks of Parkinson’s pathology.
This approach highlights best practices:
- Use of patient-derived cells preserves disease-relevant genetic context.
- Creation of isogenic controls isolates mutation effects.
- Differentiation protocols target the affected neuronal subtype.
- Functional assays validate disease phenotypes.
Mind Map: Workflow for iPSC-Based Neurodegenerative Disease Modeling
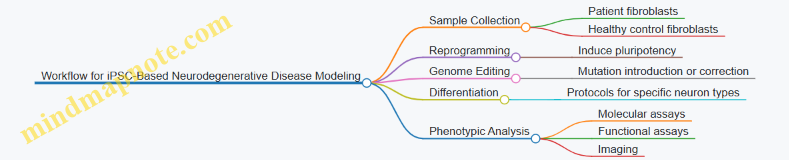
Functional Assays in Neurodegenerative Models
- Electrophysiology: Measures neuronal firing and synaptic activity.
- Calcium Imaging: Tracks intracellular calcium dynamics reflecting neuronal signaling.
- Mitochondrial Health: Assays for membrane potential and reactive oxygen species.
- Protein Aggregation: Detection of disease-related aggregates, e.g., alpha-synuclein in Parkinson’s.
- Cell Viability: Quantifies neuron survival under stress conditions.
Example: Engineering Glial Cells to Study Alzheimer’s Disease
Alzheimer’s disease involves not only neurons but also glial cells like astrocytes and microglia. Researchers differentiate iPSCs into astrocytes carrying the APOE4 allele, a risk factor for Alzheimer’s. They observe altered inflammatory responses and impaired support for neurons. By using CRISPR to correct APOE4 to APOE3, they demonstrate rescue of these phenotypes, confirming the allele’s role.
This example underscores the importance of engineering multiple cell types to capture disease complexity.
Challenges and Considerations
- Differentiation protocols vary in efficiency and purity; optimizing conditions is critical.
- Genetic background differences can confound results; isogenic controls help mitigate this.
- 3D cultures better mimic in vivo environments but are more complex to maintain and analyze.
- Off-target effects of genome editing require thorough validation.
In summary, cellular engineering enables detailed modeling of neurodegenerative diseases by combining patient-derived cells, precise genome editing, and specialized differentiation protocols. These models provide platforms to dissect disease mechanisms and test interventions with cellular context that reflects human pathology.
12.5 Integrated Example: From Design to Validation of a Therapeutic Genome Edit
This section walks through a practical example of designing, executing, and validating a therapeutic genome edit aimed at correcting a mutation in the HBB gene responsible for sickle cell disease (SCD). The goal is to replace the pathogenic allele with a corrected sequence in hematopoietic stem cells (HSCs) using CRISPR-Cas9 and homology-directed repair (HDR).
Step 1: Defining the Objective and Target
The mutation causing SCD is a single nucleotide substitution (Glu6Val) in the HBB gene. The therapeutic objective is to correct this mutation in patient-derived HSCs ex vivo, then reintroduce the edited cells.
Mind map of initial considerations:
- Therapeutic Objective
- Correct Glu6Val mutation in HBB
- Maintain HSC viability and functionality
- Target Selection
- Exon 1 of HBB gene
- Mutation site precision
- Editing Strategy
- CRISPR-Cas9 nuclease
- HDR template for correction
- Delivery Method
- Electroporation of RNP complex + ssODN
- Validation
- Genotyping
- Functional assays
Step 2: Guide RNA Design and HDR Template
Designing an sgRNA requires balancing on-target efficiency and minimizing off-target effects. Using computational tools, an sgRNA targeting near the mutation site is selected with a PAM sequence (NGG) close enough to enable efficient HDR.
The HDR template is a single-stranded oligodeoxynucleotide (ssODN) containing the corrected base and silent mutations to prevent re-cutting.
Example sgRNA sequence: 5’-GAGTCTGCCGTTACTGCCCT-3’
HDR template snippet (with corrected base in brackets):
...CTGACTCCTGAGGAGAAGTCTGCCGTTACTGCCCTG...
Silent mutations are introduced at PAM or seed regions to avoid Cas9 re-cutting after repair.
Mind map for design:
- sgRNA Design
- Target site near mutation
- High specificity score
- Low predicted off-targets
- HDR Template
- Corrected nucleotide
- Silent mutations to block re-cutting
- Homology arms (~60-90 nt each side)
Step 3: Construct Assembly and Preparation
The CRISPR-Cas9 ribonucleoprotein (RNP) complex is assembled by combining purified Cas9 protein with the synthetic sgRNA. The HDR template is synthesized as a chemically modified ssODN to improve stability.
Best practice: Prepare fresh RNP complexes immediately before electroporation to maximize activity.
Step 4: Delivery into Hematopoietic Stem Cells
Electroporation is chosen for delivering the RNP and HDR template into HSCs due to its efficiency and minimal genomic integration risk.
Key parameters:
- Cell density: 1-2 million cells per reaction
- Electroporation buffer optimized for HSC viability
- Pulse conditions fine-tuned to balance editing efficiency and cell survival
Example: Using a Lonza 4D-Nucleofector with program optimized for CD34+ cells.
Step 5: Post-Editing Culture and Expansion
Edited cells are cultured in stem cell-supportive media with cytokines (e.g., SCF, TPO, FLT3L) to maintain stemness and allow recovery.
Best practice: Minimize culture time to reduce differentiation and maintain engraftment potential.
Step 6: Validation of Editing
Genotyping:
- PCR amplification of target locus
- Sanger sequencing or next-generation sequencing (NGS) to quantify HDR and indel frequencies
Functional assays:
- Differentiation into erythroid lineage
- Hemoglobin electrophoresis to detect corrected hemoglobin
- Cell viability and proliferation assays
Mind map for validation:
- Molecular Validation
- PCR and sequencing
- Quantify HDR vs NHEJ
- Phenotypic Validation
- Differentiation assays
- Protein expression analysis
- Safety Assessment
- Off-target analysis
- Cell viability
Step 7: Off-Target Analysis and Safety Checks
Off-target sites predicted computationally are PCR-amplified and sequenced to assess unintended edits.
Best practice: Use high-fidelity Cas9 variants if off-targets are significant.
Summary Mind Map: End-to-End Therapeutic Genome Editing Workflow
This example illustrates how each step connects logically and practically, ensuring that the therapeutic genome edit is precise, efficient, and safe. The integration of design, execution, and validation is essential to move from concept to clinical application.